Manual
Contents
1. Introduction
2. Web Interface
[show]3. Standalone Program
[show]ORFanFinder is a tool that allows users to find, for a queried genome, ORFans at varying phylogenetic depths and determine the ages of a protein-coding gene based on to which phyletic level it is restricted ...


The web interface allows users to receive ORFan information about their genomes using our server, and browse the relevant information in a visual format via our website.
In order to use ORFanFinder you are required to provide the following information. First is the BLAST output file in tabular format (use "-outfmt 6" when running blast, see here for an example; also change the -max_target_seqs parameter to a large number, see below for an example); or the protein FASTA sequences of your query (a full genome or partial genome). Second, you need to have the NCBI taxonomy ID for the genome that you are querying. This can be found by using the Taxonomy Lookup tool on the right of the Submit page (see details below) or going to NCBI's Taxonomy website.
blastp -query NC_000913.faa -db nr -outfmt 6 -max_target_seqs 1000000 -evalue 1e-3 -num_threads 8 -out ecoli.faa.nr.out
The Submit a Job page allows you to submit your query in order to find ORFans. Fill out the form as follows:


If you do not know the taxonomy ID of your genome, you can do a lookup here. You can type in any portion of the NCBI name in order to do the lookup. This means that using the species name would be valid entries. Results are sorted first by those that start with what you have typed, and alphabetically from there. When you click on a result, the taxonomy ID will be placed in the appropriate box in the submit form.
You can also search NCBI website to get the taxonomy ID. Check this PDF for instruction.

Once your job has finished, you will be taken to a page that looks like the below. This is a graphical representation of the distribution of your results. Each level of ORFan is represented by a bar that shows how many of your proteins fell into that category. Clicking on the bar will take you the table, filtering by that ORFan category. You can also change the Display Type dropdown to "Table" to move to the table view.
You can also download a tab-delimited excel format file with detailed info to your local computer; here is an example

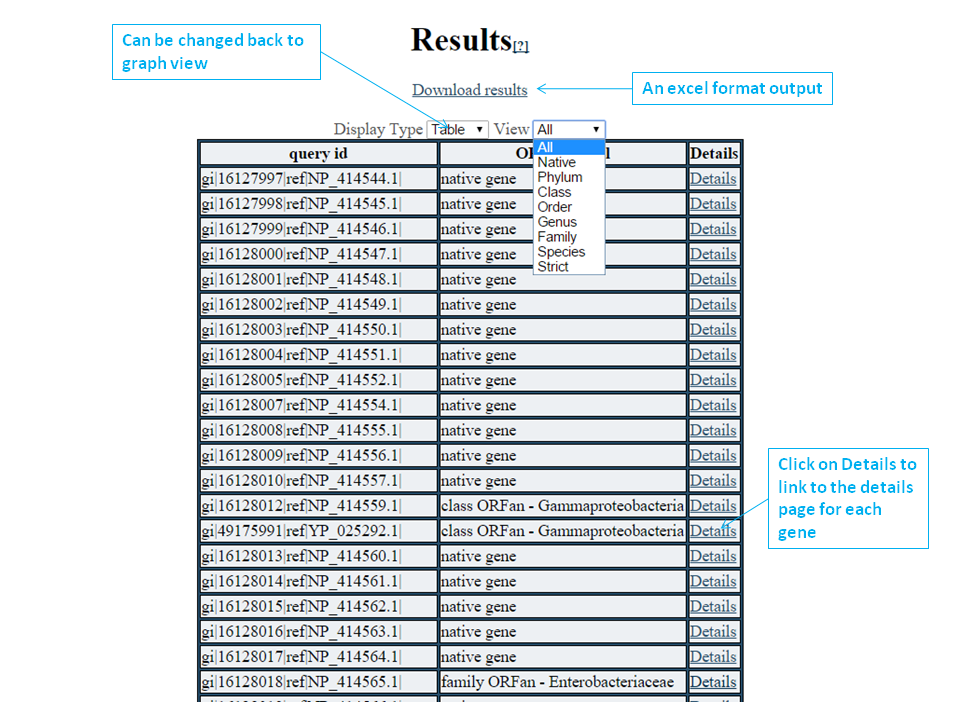
By switching to a tabular view, you can see which of your proteins fall into which ORFan categories. These will be displayed in the same order that they were in when you submitted your FASTA information. You can filter your results to a particular category by changing the View dropdown. Clicking on details will bring you to the details page for that protein.
When you view the details page, you get information that is specific to the protein that you are viewing. The hit tree provides a view of what ranks were hit by the blast, and how many to each. You can close or open the entire tree by hitting the appropriate button. A table is also presented to show how many ranks of the total ranks at that level were hit. Finally, a table with the BLAST results for this protein is displayed, and it shows the taxonomy lineage info of the subject protein in the NCBI-nr DB.

You can see jobs that you have run recently by going to the recent results page. Click on an ID link to be taken to that results page. Please note that you must have cookies enabled in order to use this feature.
Occasionally the program will not turn up any results, leading to the screen that reads "No ORFans Found". This is generally not an errorm but rather based on the fact that all genes were not catagorized as ORFans. For example, if the genes could possibly be horizontally transferred. This case generally only happens with small queries. If you believe that this is in error, please contact us through email.
Go to the download page to download source code and related files. The source is platform independant, although you will need to compile it.
Suppose you are at your home folder:wget cys.bios.niu.edu/orfanfinder/ORFanFinder.tar.gz
In order to compile, you will need a C++11 compatible compiler. Compilers such as newer versions of GCC and Clang come with this comatibility. The compilation process follows the standard GNU make process. It is recommended that you run the commands from a directory outside of the downloaded folder. A potential command sequence could look like the following:
tar zxf ORFanFinder.tar.gz
cd ORFanFinder
sudo ./configure #: if you are a root/superuser
./configure --prefix=$HOME/ORFanFinder/ #: if you are NOT a root/superuser, this will install the program in $HOME/ORFanFinder/ folder
make
make install
make db
This will result in an executable called ORFanFinder that can be run on your system. By default it is placed in the bin folder in your path, but this can be configured with the --prefix option when running configure.
There are several files that you will need prior to running ORFanFinder. The first is your BLAST file that you get from querying your genome against your database. Make certain that it is in tabular format (i.e. -outfmt 6 when running blastp). Second, you need a list of all gene IDs from the FASTA. You can generate these by passing your FASTA file to the extractIdsFromFasta command, provided when ORFanFinder was installed. The third thing that you need is the taxonomy ID of your genome from NCBI. Fourth, you need a nodes file that describes the level and parent of each taxonomy ID. If you wish to use the default data set, this file is included in the download, and is named "nodes", in the inputData folder. This file can be created from NCBI's nodes.dmp with the following command:
sed 's/\t[|]\t/\t/g' nodes.dmp | cut -f1,2,3
The same applies to the names file, if you wish to use it.
sed 's/\t[|]\t/\t/g' names.dmp | cut -f1,2
If you wish to use your own data set, you must make this file in this format. The last file that you need is a database file generated by the program ORFanDbFormat, which is provided with ORFanFinder. If you are simply looking to use one of the databases that we use on the website, running 'make db' in the build directory will compile several databases, including nr, and put them into the databases directory. Otherwise ORFanDbFormat will require an input file that contains each protein in the BLAST database and its taxonomy ID, being tab-delimited.
To run ORFanFinder, the general format is:
ORFanFinder -query blast_file -id id_list -tax taxID -nodes nodes_file -db database_file -out out_file
Output is through the provided output file argument. Assuming that you are on Unix and using the default file nodes, a run may look like this:
ORFanFinder -queury ecoliMG1655.bl -id idList -tax 511145 -nodes nodes -db nr.hdb -out ecoliMG1655.out
By supplying a list of names, the program will display the extra data that the webserver uses to construct the hit table and the hit tree.
ORFanFinder -query ecoliMG1655.bl -id idList -tax 511145 -nodes nodes -db nr.db names -out ecoliMG1655.out
The results come in a tab delimited file, where the first column is the protein ID, and the second column is the ORFan level. If you provided the names file, extra columns wil be added on. Each of these fields is a phylogenetic level; the first section is in brackets, giving the name and number of hits in that rank. After that are each of the hits in that rank, with that rank's parent in parentheses. Check here for an example. It is delivered through the standard output.