Email us for technical support
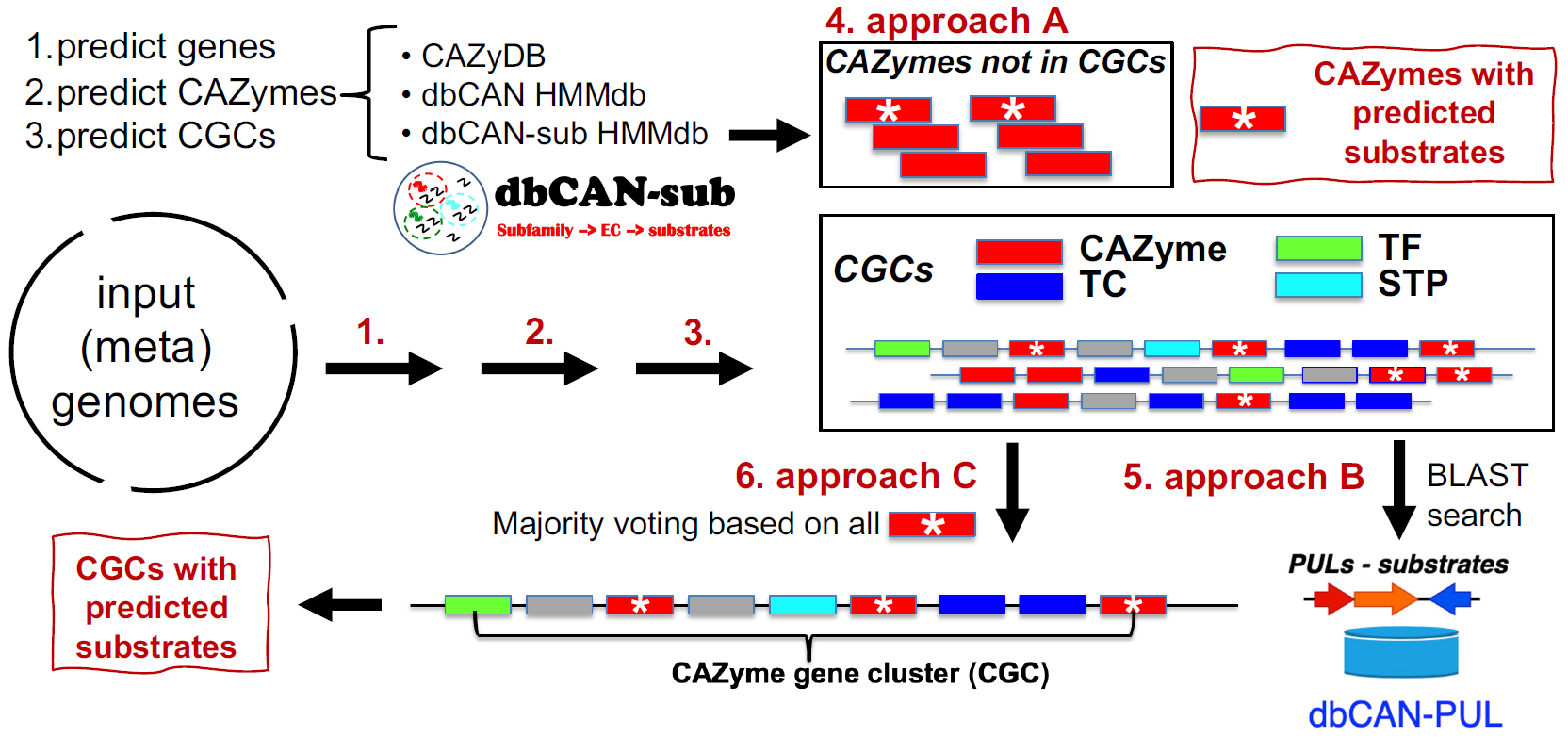
Overall design of dbCAN3 server
How accurate is our substrate prediction?
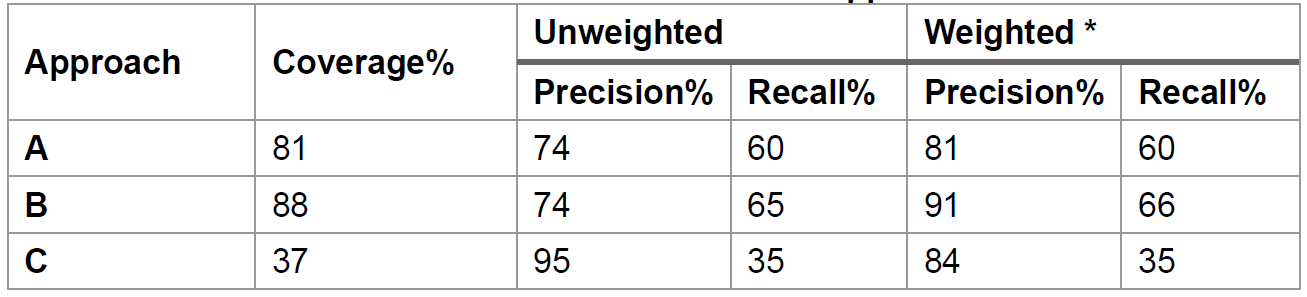
We benchmarked the three approaches of substrate prediction using experimentally characterized PUL data from
dbCAN-PUL. Three metrics are used: coverage, precision, and recall. The details are described in our
dbCAN-seq paper and
dbCAN3 paper, but briefly:
- Approach A (dbCAN-sub HMMdb): For a query CAZyme, dbCAN3 can predict the substrate based on subfamily annotation in dbCAN-sub. This approach has weighted precision = 81%, weighted recall = 60%, and coverage = 81%.
- Approach B (dbCAN-PUL search): For each CGC, dbCAN3 can predict the substrate based on BLAST search against PULs in dbCAN-PUL. This approach has a coverage = 88%, weighted precision = 91% and weighted recall = 66%.
- Approach C (dbCAN-sub majority voting): For each CGC with multiple CAZymes, using a simple majority voting rule, a substrate assignment for the CGC can be inferred by considering all the CAZymes in the CGC and their substrates inferred by approach A. This approach has a weigthed precision = 84%, weighted recall = 35%, and coverage = 37%.
- Therefore, the approach B (dbCAN-PUL search) has the best performance (highest coverage, weighted precision, and recall), followed by the approach A (dbCAN-sub search for CAZyme substrate prediction) (Table 1). Approach C (dbCAN-sub majority voting) suffers from low coverage and recall.
Tool Info
- DIAMOND: E-Value < 1e-102, hits per query (-k) = 1.
- HMMER(dbCAN): E-Value < 1e-15, coverage > 0.35.
- HMMER(dbCAN-sub): E-Value < 1e-15, coverage > 0.35.
- CGC-Finder: Distance = 2, signature genes = CAZyme+TC. DIAMOND for TC and TF: E Value = 1e-10, hits per query (-k) = 1. CAZymes are defined as those predicted by at least 2 tools.
- Prodigal: Defaults.
- CGC substrate prediction based on dbCAN-PUL search: unique genes homologous >= 2, bitscore >= 50, total signature pairs >= 2 and CAZyme signature pair >= 1.
- CGC substrate prediction based on eCAMI-subfam: major voting score >= 2.
Annotate Protein Sequences
-
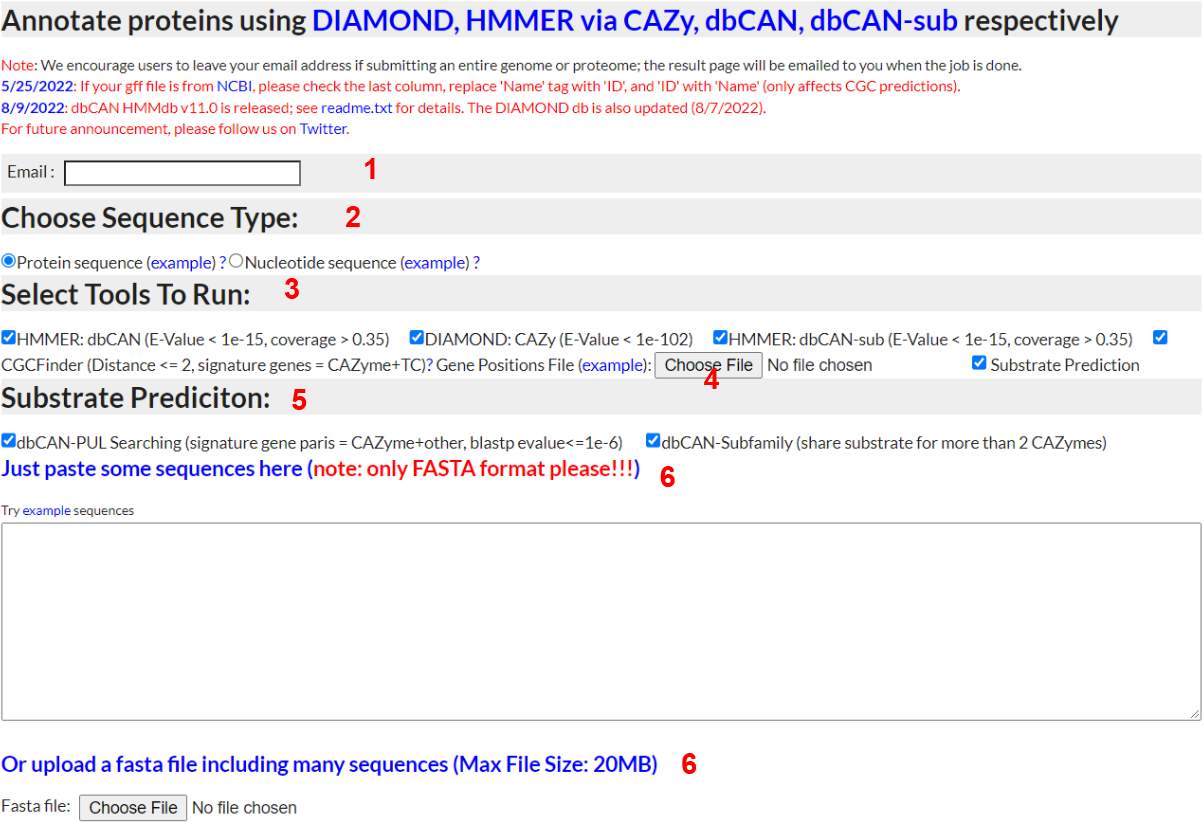
1) Email
- - Input a valid email address and dbCAN meta server will email you when your job completes
2) Sequence Type
- - To annotate a protein sequence, select "Protein sequence"; to try out the example, right click and save as the file to your computer and then upload (see 5 below)
-
3) Select tools to run
- - Selecting CGC-Finder will display the gene position file upload button; you must upload a gene position file (example provided) to have CGC-Finder predict CAZyme gene clusters (CGCs).
- - CGCs are defined as genomic regions containing at least one CAZyme gene, one transporter/TC gene (predicted by searching against the TCDB) and one transcription factor/TF gene (predicted by searching against the collectf DB, the RegulonDB, and the DBTBS. The rational is that CAZymes often work together with each other and with other important genes (e.g. TFs, sugar transporters) to synergistically degrade or synthesize various highly complex carbohydrates. See our paper for why CGCs are interesting to identify.
-
4) Gene Positions File
- - If you chose to run CGC-Finder, you must upload a GFF or BED format file (see here for an BED example) that contains position data on each gene you upload. Gene ID's used in the FASTA file must exactly match those in the BED/GFF file. If using a GFF file, only rows with 'CDS' in the type column will be considered. If using a GFF file, gene ID's should be in the notes column with the Name tag; if no Name tag is present then the gene ID should be in the ID tag.
-
5) Method to predict substrate
- -dbCAN-PUL search. his approach is based on the BLAST comparison of protein sequences of the query CGCs against the protein sequences of the 612 PULs of dbCAN-PUL. To improve the confient of the CGC predict, all the CGC hits are sorted by the summed bit-scores of each genes. In addtion, it requires at least one CAZyme match plus at less one match from one of the other 4 signature genes.
-
-eCAMI sub-family. This pproach is based on the inspection of the eCAMI subfamily annotated substrates of all component CAZymes in the CGC. The substate of CGC is dependent on the substrate of CAZymes in the CGC. It requires at less two shared two CAZyme substrate to obtain a higher confident substrate for the CGC. The final substrate is decided as the most common shared CAZyme substrate
6) Sequence Input
- - You can either paste protein sequences into the textbox or upload a file containing protein sequences. In either case, the sequences must be in FASTA format.
Annotate Nucleotide Sequences
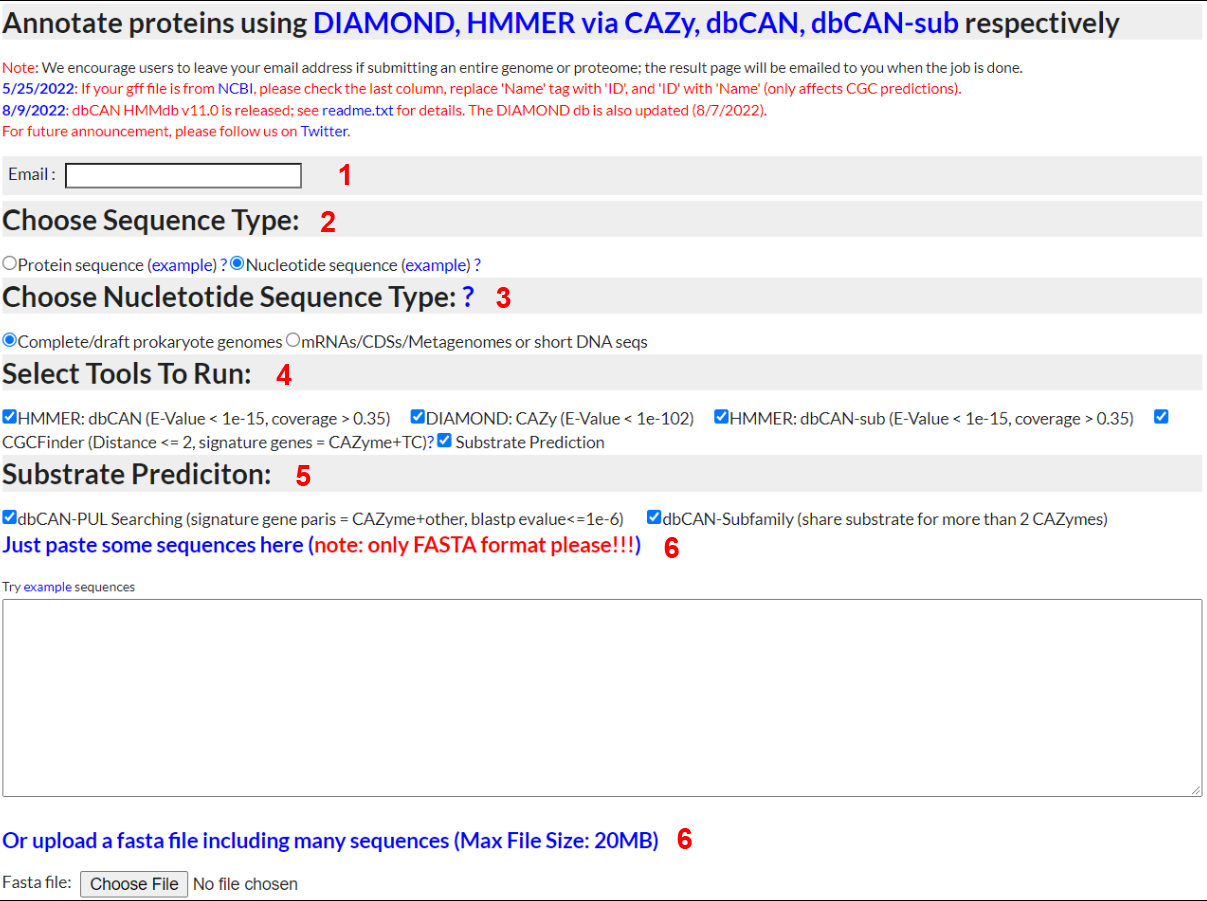
-
1) Email
- - Input a valid email address and dbCAN meta server will email you when your job completes
-
2) Sequence Type
- - To annotate a nucleotide sequence, select "Nucleotide sequence"; to try out the example, right click and save as the file to your computer and then upload (see 5 below)
3) Nucleotide Sequence Type
- - To annotate prokaryote genomes, select "Complete/draft prokaryote genomes". To annotate metagenomes, select "Metagenomes". FragGeneScan is used for gene prediction in
Metagenomes, and Prodigal is used for gene prediction in prokaryote genomes. If you have an eukaryotic genome, please run gene finding softwares elsewhere (e.g. MAKER) and then submit protein sequences.
-
4) Run CGC-Finder and predict substrate
- - Select "Yes" to have CGC-Finder predict CAZyme gene clusters. Select "Yes" to choose method to predict substrate of CGC.
-
5) Method to predict substrate
- -dbCAN-PUL search. his approach is based on the BLAST comparison of protein sequences of the query CGCs against the protein sequences of the 612 PULs of dbCAN-PUL. To improve the confient of the CGC predict, all the CGC hits are sorted by the summed bit-scores of each genes. In addtion, it requires at least one CAZyme match plus at less one match from one of the other 4 signature genes.
-
-eCAMI sub-family. This pproach is based on the inspection of the eCAMI subfamily annotated substrates of all component CAZymes in the CGC. The substate of CGC is dependent on the substrate of CAZymes in the CGC. It requires at less two shared two CAZyme substrate to obtain a higher confident substrate for the CGC. The final substrate is decided as the most common shared CAZyme substrate
6) Sequence Input
- - You can either paste DNA sequences into the textbox or upload a file containing DNA sequences. In either case, the sequences must be in FASTA format.
Result page
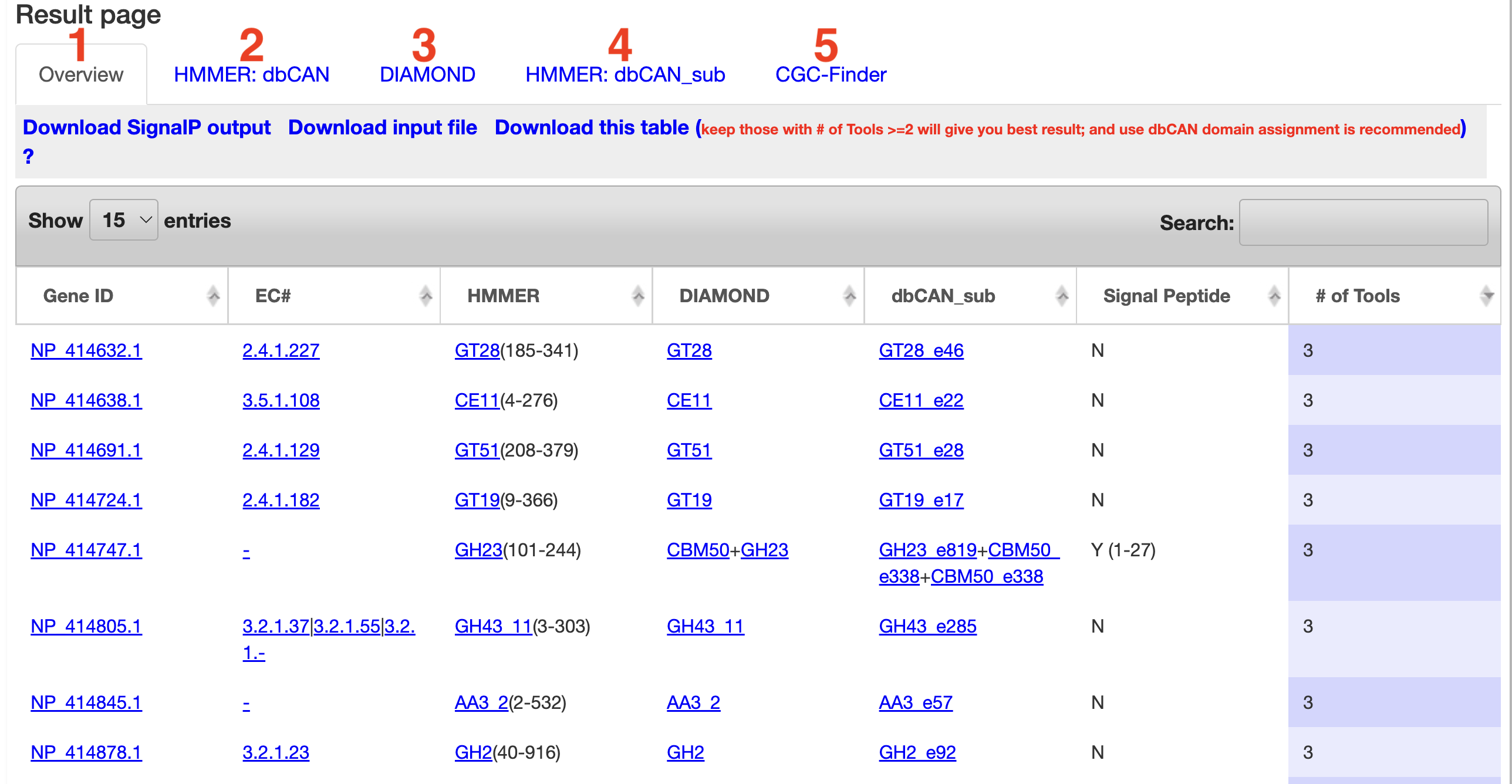
-
1) Overview
- - This tab shows an overview of all the tools run. Each annotated protein is displayed along with which tools annotated it and what CAZy family they were annotated in. Each CAZy family is also a link
to the CAZy web page for the appropriate family. Along with this,
signal peptide predictions are displayed. The full signalp output is avaliable for download at the top of the tab. The table is also available for download along with the gene predictions (if a nucleotide
sequence was uploaded).
- - The # of Tools can be sorted and proteins predicted by more tools are more reliable CAZyme candidates. Our benchmark analysis suggests keeping proteins found by >=2 tools can give the best CAZome annotation performance.
- -About compasiron of the three tools: se have also systematically compared the outputs of the three tools against the CAZy pre-annotated CAZomes (i.e., as the gold standard sets) of three bacterial genomes and three eukaryotic genomes.
The accuracy is calculated as an F-score = 2 × (Recall × Precision)/(Recall + Precision) for the three methods on each examined genome, following the method presented in our dbCAN-seq paper and PlantCAZyme paper. We removed unclassified CAZymes (e.g. GH0) and families not in the PPR library when calculating F-scores.
- - Advantage of HMMER search against dbCAN: However, the F-score calculation only considered whether a protein is found by any of the three tools. It did not consider if the protein is assigned to the correct family or families, if the protein has multiple CAZyme domains, and where the domain boundaries are. The below Figure shows two example CAZyme proteins found by all the three tools. Both proteins have multiple CAZyme domains according to dbCAN annotation (Figure A). According to HMMER+dbCAN output (Figure C), NP_414632.1 is annotated as GT28(185-341) and NP_414638.1 as CE11(4-276). According to both HMMER+dbCAN-sub output and DIAMOND+CAZy output, NP_414632.1 is annotated as GT28 in DIAMOND, GT28_e46 in HMMER(dbCAN-sub).
It should be mentioned that DIAMOND+CAZy has a much higher risk than the other two tools to give wrong CAZyme family annotation. For example, if a query protein only has a GT5 domain and has AAD30251.1 as its best CAZy hit, transferring the family assignment of AAD30251.1 (GT5+CBM53) to the query would be wrong (as no CBM53 in the query). However, such mistakes will not happen in HMMER and eCAMI searches, as they are conserved domain and motif-based methods.
- - The Gene IDs found by HMMER and DIAMOND are clickable in Overview table, which will open the protein domain display page.
-
2) HMMER: dbCAN
- - This tab displays the results of the HMMER run versus the dbCAN database. The full output is avaliable for download via a link at the top of the tab.
3) DIAMOND: CAZy
- - This tab displays the results of the DIAMOND blast versus the CAZy database. The full output is avaliable for download via a link at the top of the tab.
-
4) HMMER: dbCAN-sub
- - This tab displays the results of the HMMER run versus the dbCAN-sub database. The full output is avaliable for download via a link at the top of the tab.
5) CGC-Finder and substrate
- 1) CGC and substrate result table
This tab dispalys the output of CGC-Finder, if the user chose to run CGC-Finder.
- 2) CGC result download links
Several files are avaliable for download at the top of this tab. The full input and output files for CGC-Finder are avaliable, along with the full DIAMOND outputs that were used to annotate genes as TCs (transporters) or TFs (transcription factors). - - CGCs are defined as genomic regions containing at least one CAZyme gene, one transporter/TC gene (predicted by searching against the TCDB) and one transcription factor/TF gene (predicted by searching against the collectf DB, the RegulonDB, and the DBTBS. The rational is that CAZymes often work together with each other and with other important genes (e.g. TFs, sugar transporters) to synergistically degrade or synthesize various highly complex carbohydrates.
- 3) Rerun CGC-Finder:
At the bottom of the CGC-Finder tab, users can choose to rerun CGC-Finder with customized settings. The distance setting is the maximum number of non-signature genes allowed between signature genes. The signature genes setting is which signature genes are required to be in a cluster in order for the cluster to be annotated as a CGC. The CGC-Finder rerun is superfast, and the page will return back to Overview; just clicking on the CGC-Finder tab to view the new CGC result - 4) Rerun substrate prediction with more parameters:
At the bottom of the CGC-Finder tab, users can choose to rerun substrate prediciton with customized parameters including three parts: Part1 is the parameters for Blastp defining the homolgous between genes; Part2 is the parameters for dbCAN-PUL search defining the homologous bewteen CGC; Parts3 is the parameters for dbCAN-sub family, related to major voting. This part is fast, and the page will return back to Overview; just clicking on the CGC-Finder tab to view the new substrate result. If you want to obtain the substrate prediction of dbCAN-sub family, please check the box using dbCAN-sub to annotate the CAZyme when runing dbCAN3.-
- The individual CGC and substrate page:
clicking on the CGC ID will open a new page shown as following 1) CGC gene composition diagram; and interactive interface to show the information of CGC. 2) Gene composition table. 3) syntenic diagram of the best hit in dbCAN-PUL showing the comparion of two homologous CGC in detail. 4) blastp results showing all the gene homologous hits in dbCAN-PUL. 4) dbCAN-sub family table showing all the substate of CAZyme within the CGC.