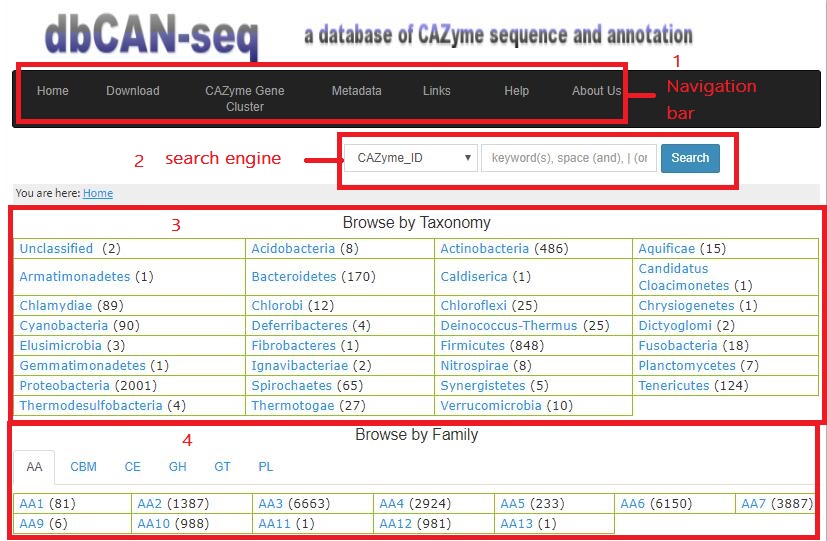
- Index There are four ways to reach the annotation page for each individual protein: (i) browse by taxonomy or (ii) by CAZy family, or (iii) search using key word or (iv) browse by metadata. In the home page (figure below), the number in the Browse by Taxonomy section (in parentheses) is the number of genomes; the number in the Browse by Family section (in parentheses) is the number of CAZyme proteins.
* Browse page click to see this sample page.
-
The navigation contains Home, Download, CAZyme Gene Cluster, Metadata, Links, Help and About Us.
- 1.1 Download The CAZyme sequence and annotation data are available in the download page.
- 1.2 CAZyme Gene Cluster
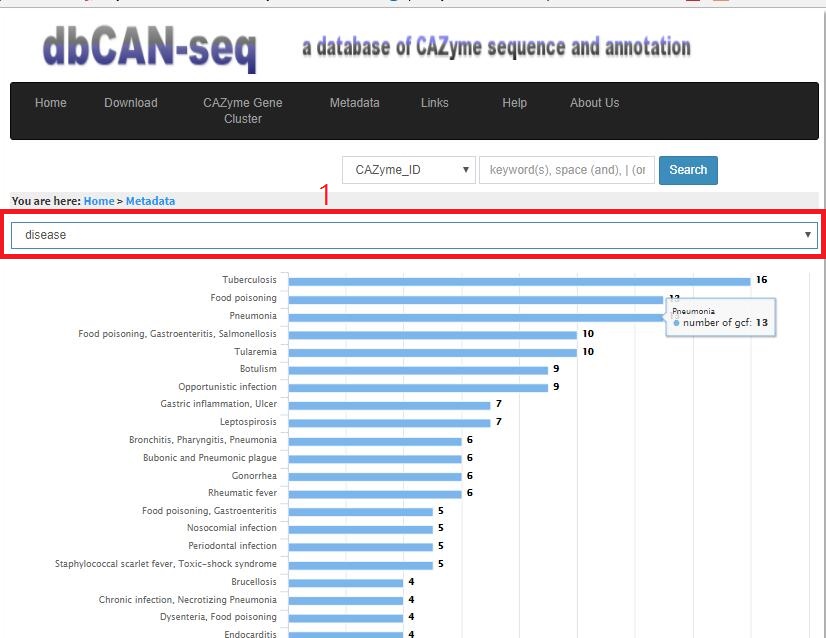
- 1.3 Metadata Metadata are the macroscopical terms describing different biological properties. Here we downloaded all the bacteria's species metadata from JGI's Integrated Microbial Genomes (IMG) database. We then extracted genomes having at least one metadata property in our dbCAN-seq database. The properties are disease, ecosystem, ecosystem category, ecosystem type, habitat, metabolism, motility, oxygen_requirement, ph, phenotype, salinity, sample body site, sample body subsite, specific ecosystem, temperature range. All these features could be selected from a pull-down menu. When one feature is selected (e.g., disease in the red box), a bar chart will be displayed with y axis showing the different diseases and the bar height showing the number of genomes/GCFs in that disease. The bars are sorted according to height. Clicking one bar will lead to a new page with another bar chart showing the genomes/GCFs in that bar (below).
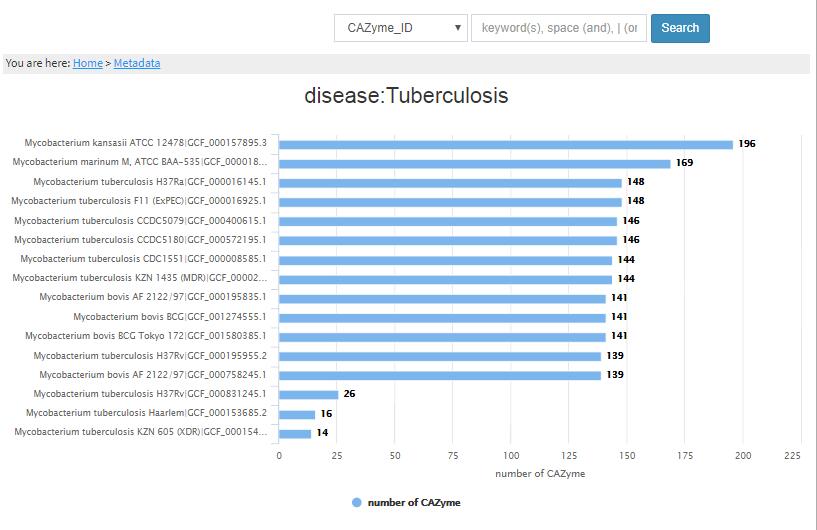
- 1.3.1 Metadata-property This bar chart is sorted according to height too. Each bar represents one GCF/genome, with the height proportional to the number of CAZymes found in the genome. Clicking one of them, we will go to the genome page.
Introduction:
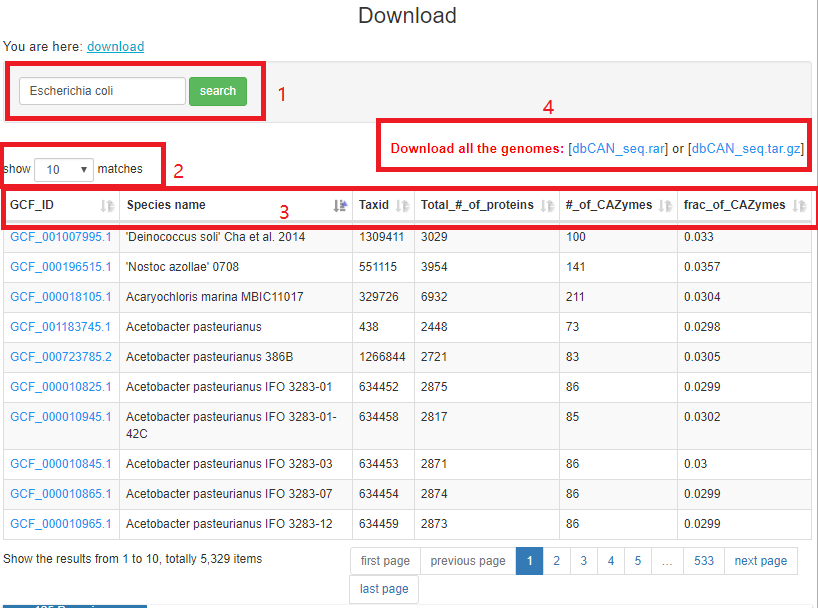
The download page has a searchable table with all the 5,329 genomes. Each row of the table corresponds to a genome with a download link to a compressed tarball file. The tarball contains a FASTA sequence file of all the CAZymes in the genome, and a tab-separated file with all the annotation and location data. There is also a link to download data of all the genomes.
There are three functions in this page (red boxes in below picture):
1. You can put key words (GCF ID [NCBI genome assembly ID], species name and taxid) to search the download table
2. You can choose to show different numbers of entries
3. There are six properties shown for each genome including the fraction of CAZyme in the genome
4. You can download all the genomes file.
Click the GCF ID, you can download the tar.gz file; this tar ball contains two files:
1. GCF_ID.fasta contiains the all of the CAZymes sequence of this GCF_ID.
2. GCF_ID.txt contains the following tab-separated properties of each CAZymes:
(GCF_ID, CAZyme_ID, Product, RefSeq_ID, Start, End, Strand, CAZyme_domains, Molecular_weight, Isoelectric_point, TMHMM_num, LipoP, Predicted_EC, MetaCyc, SignalP_cleavage_site).
Introduction:
CGCs are defined as genomic regions containing at least one CAZyme gene, one transporter/TC gene (predicted by searching against the TCDB) and one transcription factor/TF gene (predicted by searching against the collectf DB, the RegulonDB, and the DBTBS. The rational is that CAZymes often work together with each other and with other important genes (e.g. TFs, sugar transporters) to synergistically degrade or synthesize various highly complex carbohydrates.
The CGC page is designed as a tool page. When a user opens the "CAZyme Gene Cluster" page, a table will be seen with all the 5,329 genomes. The table actually has 7,841 rows as one genome can have multiple RefSeq IDs (chromosomes and plasmids).
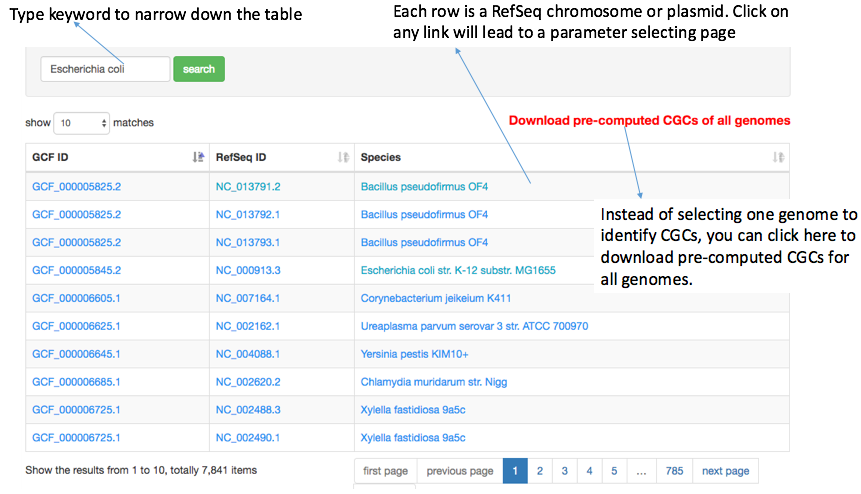
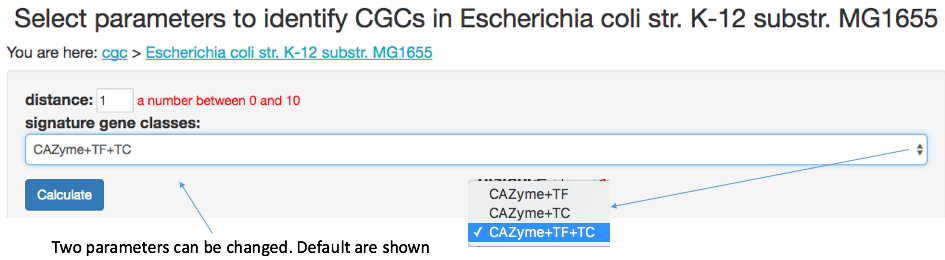
One can click on a genome to open a new page (parameter page), where two parameters need to be set: (i) distance and (ii) signature gene classes, which were already described above. After hitting on the Calculate button, the CGC-Finder python program will be called to identify CGCs in the genome. The program runs very fast: processing one genome takes a few milliseconds, and processing all the 5,329 genomes together takes < 1 minute.
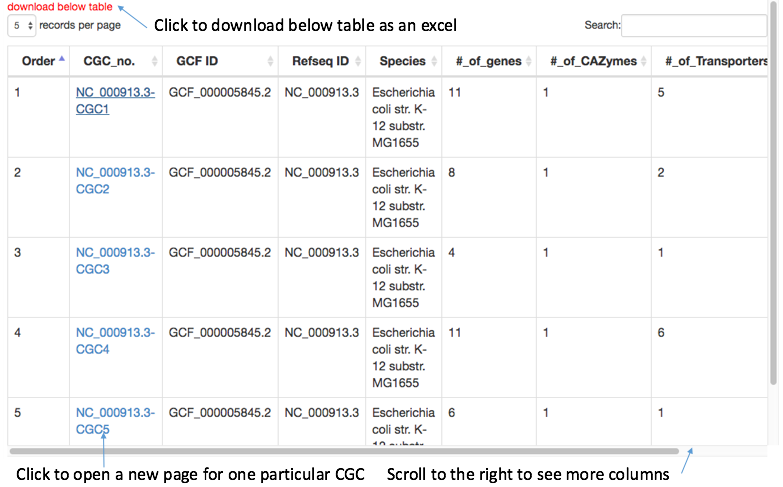
The CGC-Finder result will be printed as a table (CGC genome page) below the parameter selection section. Each row in the table is one CGC with different statistics about the CGC, such as the numbers of the three signature genes and the number of all genes (including non-signature ones).
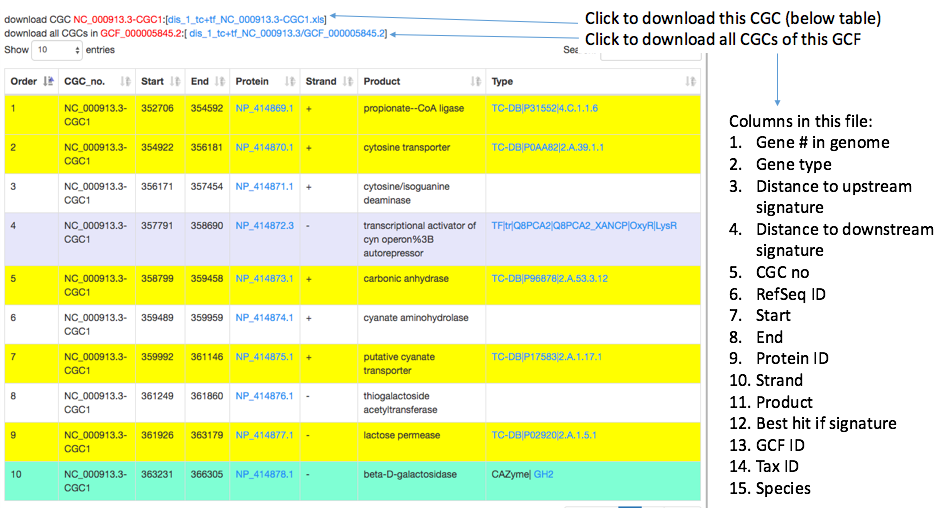
Clicking on the CGC_no will open a separate page (individual CGC page) showing: (i) the graphical representation of the genomic location of the CGC in a Jbrowser; (ii) at the bottom the detailed information about all genes in the CGC as a table, such as the genomic location, the functional description, and if signature gene and evidence. All the data tables can be downloaded by clicking on a download link above the table.
* Search Engine
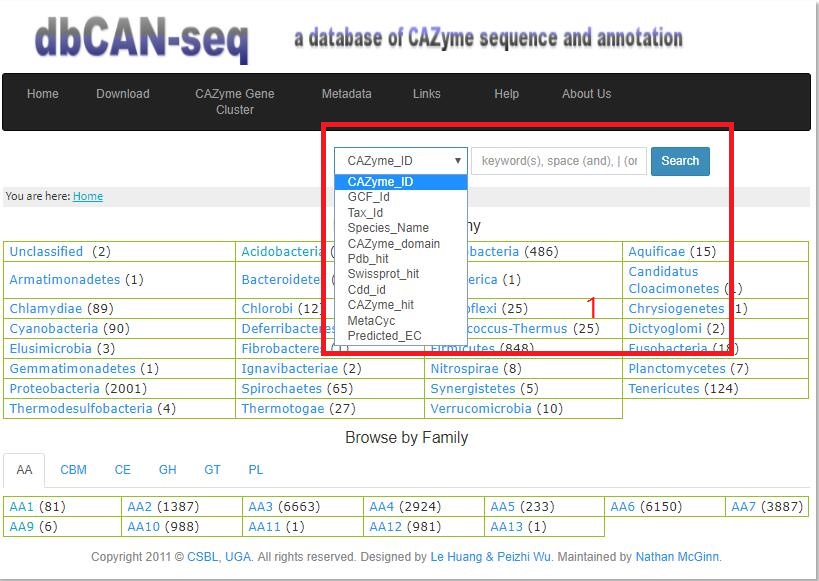
- 2 Search Engine We support keyword search of 11different data fields:
1. CAZyme ID (e.g., NP_212393.2 );
2. GCF_ID (e.g., GCF_000005825.2);
3. Tax_ID (e.g., 398511);
4. Species_Name (e.g.,Bacillus pseudofirmus OF4);
5. CAZyme_domain (e.g., CE4);
6. Pdb_hit (e.g., similarity = 20%,1LZL_A);
7. Swissprot_hit (e.g.,similarity = 20%,ETHA_MYCTU or P9WNF9 or sp|P9WNF9|ETHA_MYCTU);
8. Cdd_ID (e.g., similarity = 20%,COG2072);
9. CAZyme_hit (e.g., similarity = 20%,AHF23796.1);
10. MetaCyc (e.g., 3.2.1.8-RXN);
11. Predited_EC (e.g., 3.1.1.23);
From 1 to 5 and 10 to 11, one can type the keyword to search.
From 6 to 9, one has to specify a sequence identity value in addition to a keyword. For example, if choose to search pdb_hit, on the left an identity value, e.g., 20%, has to be selected, followed by a pdb ID. The result will be a list of proteins in the database that share > 20% identity to the pdb protein.
Our search Engine is very fast, because we use sphinx to make different page. When that page is on, the search results on that page will be obtained rather than all of the search results obtained no matter the page is on or not.
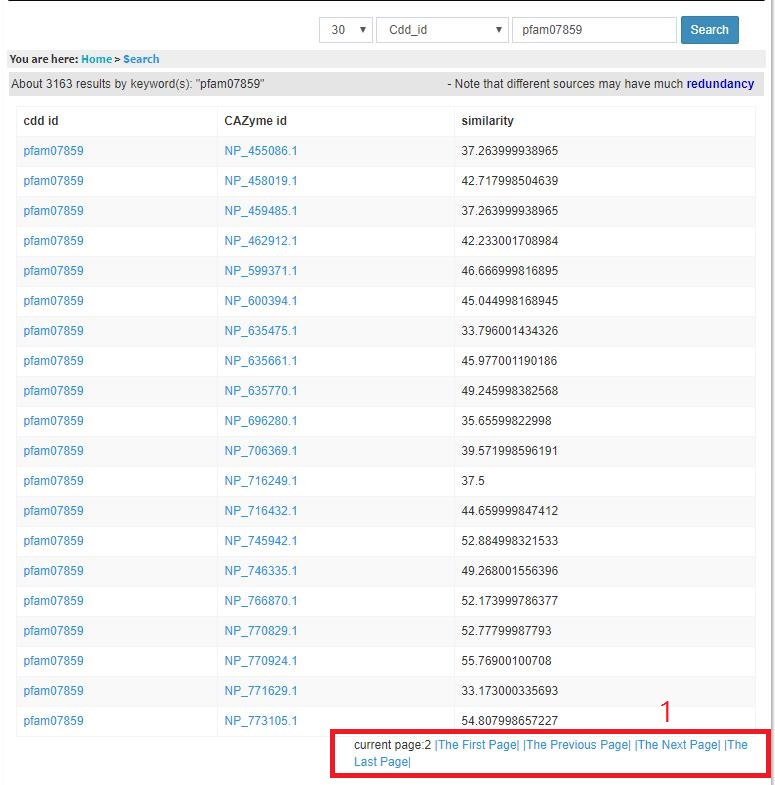
Take Cdd_id(e.g.,similarity = 30%,pfam07859) for example,
this is page 2 of the search result of cdd_id query pfam07859.
When we click "The Next Page/The Previous Page", then we will get related result of this page.
* Browse page click here to this sample page.
- 3 Browse by Taxonomy
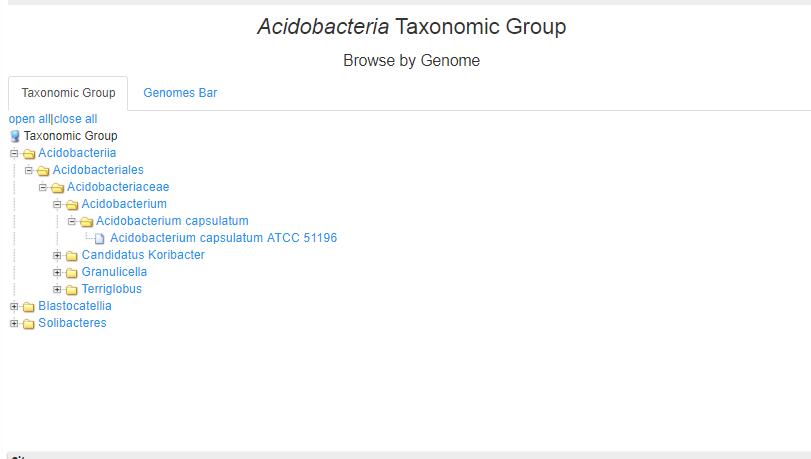
- 3.1 This page shows the 31 phyla of bacteria. Choose one of phylum,the six levels of classification (phylum, class, lineage_order, family, genus, species) are shown when choose Taxonomic Group.
- 3.2 Browse by Family Genome Bar version shows the sorted number of CAZymes of each taxnomy|gcf_id, where all of the numbers are sorted. Choose one of them, we go to the "Browse by Genome" page.
- 3.3 Genomes Bar Genomes bar shows the sorted number of CAZymes of each taxnomy|gcf_id, where all of the numbers are sorted. Choose one of them, we go to the "Browse by Genome" page.
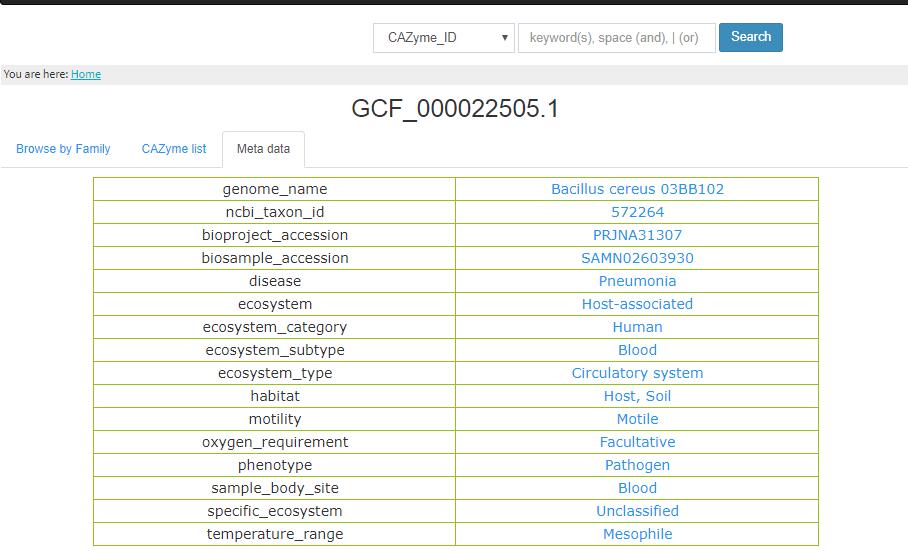
- 3.4 metadata If the genome has metadata in our database, the page will show the metadata of all lot of properties.
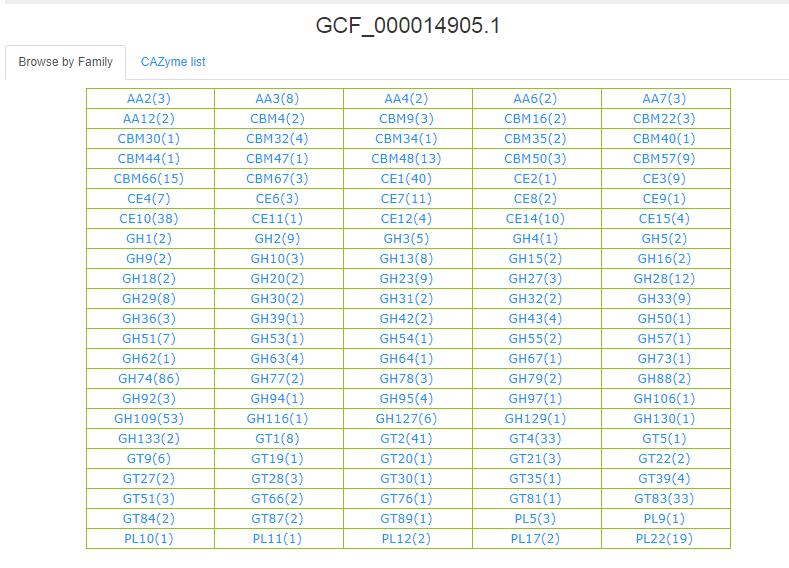
- 3.5 Browse by Genome This page has two tabs(Browse by Family, CAZyme list).
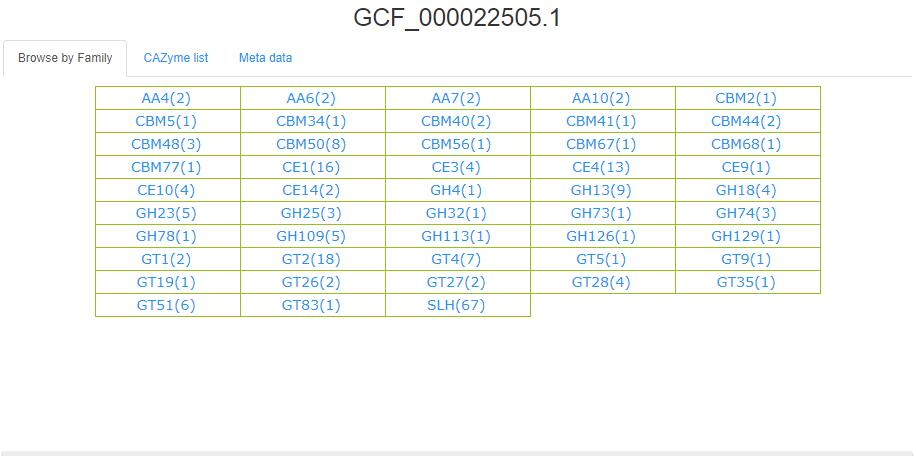
The CAZymes number of each family of this Genome are caculated and shown in the bracket behind each family name.
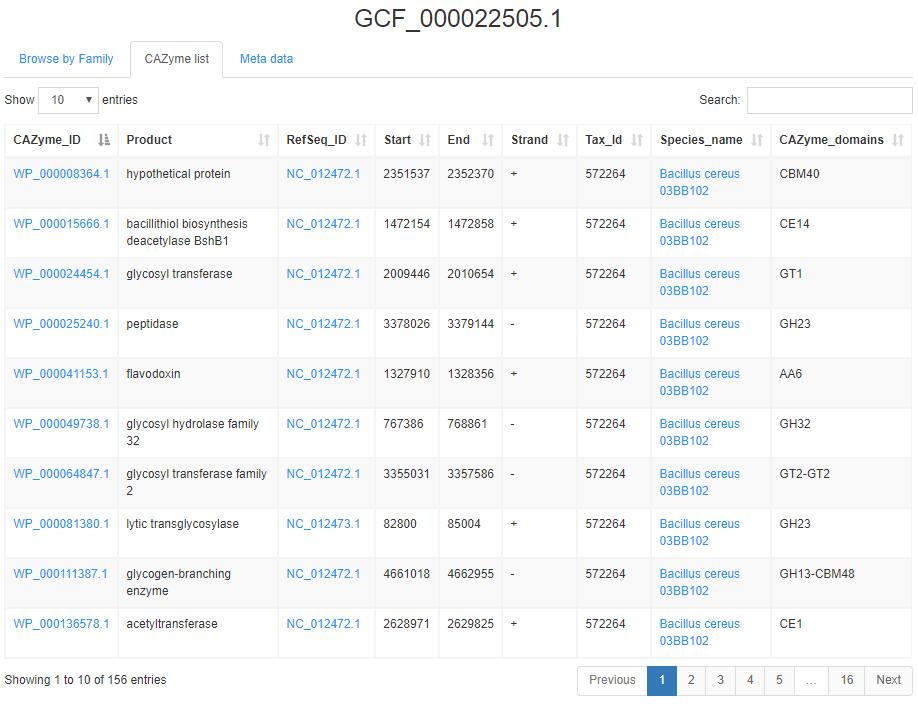
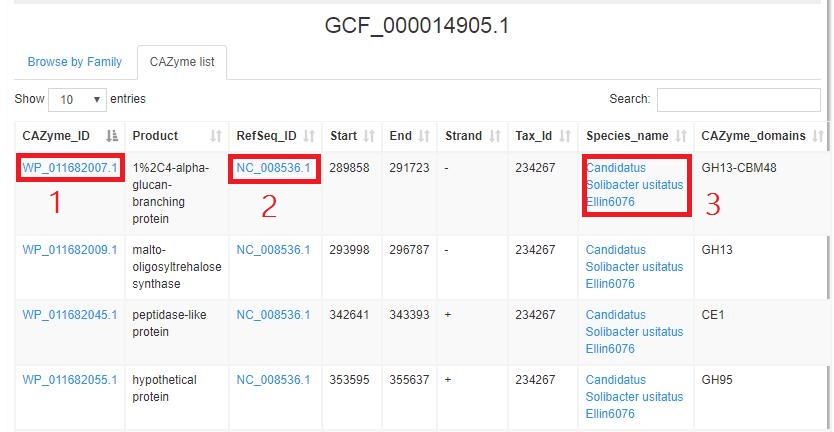
CAZyme lists (sample)shows the detailed information of CAZyme.
1.Red rectangle 1 links to the protein page (sample).
2.Red rectangle 2 links to the NCBI database(sample).
3.Red rectangle 2 links to the NCBI taxnomy page(sample).
* Protein page click here to this sample page.
- 4 Browse by Family We can choose different family from the fourthpart of index page, this families contains:
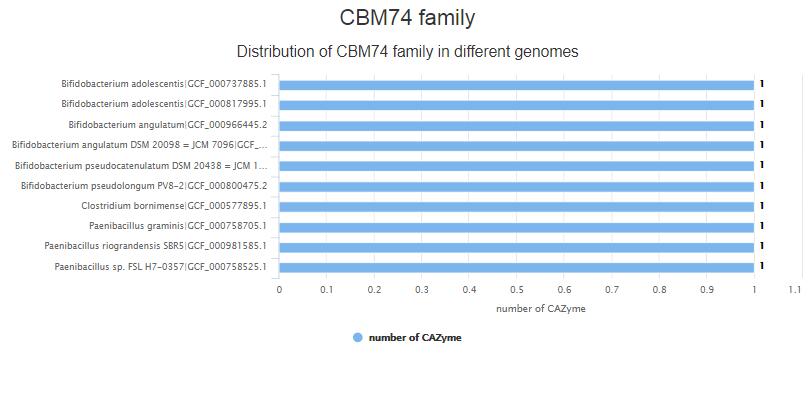
- 4.1 Family page This page contain all of species|genome of the CBM74 family, # of CAZymes of one species are caculated, the bar plot are sorted.
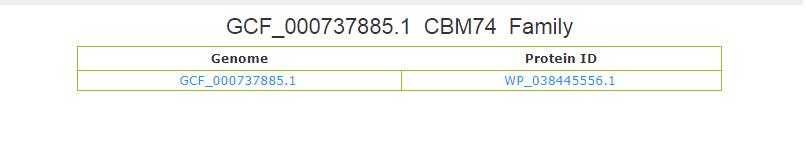
- 4.2 Genome Family page This page shows the all of the proteins of GCF_000737885.1 genome of CBM74 family.
- Auxiliary Activities (AAs) : redox enzymes that act in conjunction with CAZymes.
- Carbohydrate-Binding Module (CBMs) :a contiguous amino acid sequence within a carbohydrate-active enzyme with a discreet fold having carbohydrate-binding activity.
- Carbohydrate Esterases (CEs) : hydrolysis of carbohydrate esters.
- Glycoside Hydrolases (GHs) : hydrolysis and/or rearrangement of glycosidic bonds.
- GlycosylTransferases (GTs) : formation of glycosidic bonds.
- Polysaccharide Lyases (PLs) : non-hydrolytic cleavage of glycosidic bonds.
Choose one of them(select CMB74 as example), then go to he family page.
Chooese one of them, we go to the genome_family page.
Click one of them, we go to the protein page(protein help page).
* Protein page , click here to this sample page.
- 5 Protein Page
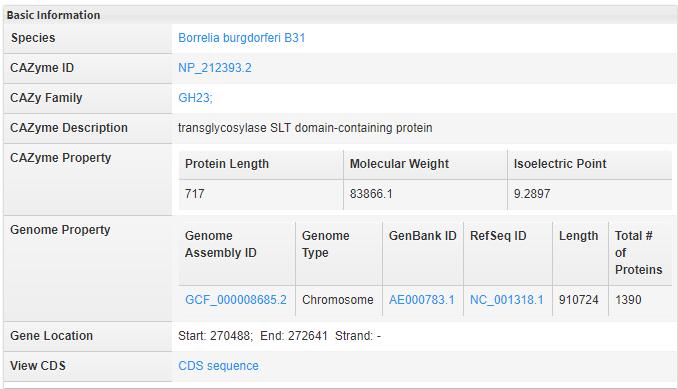
- 5.1 Basic Information The basic information descibes the property of CAZyme and its genome. All of them have the outer link to related databse.
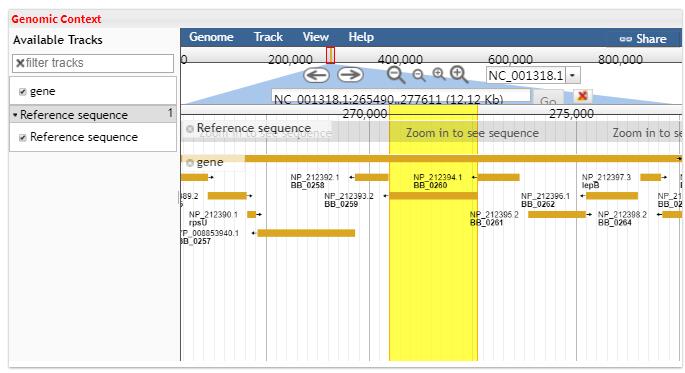
- 5.2 Genomic Context Jbrowse use the GFF3 file to label the gene location of one genome.
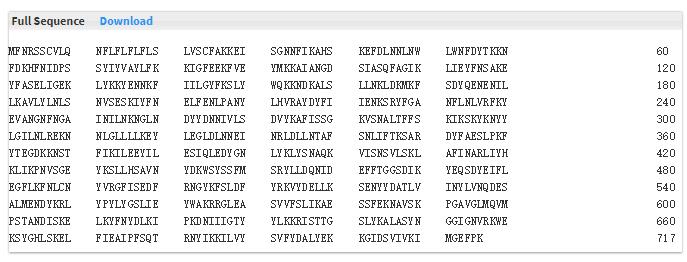
- 5.3 Full sequence We support the full sequence of protein and the download link.
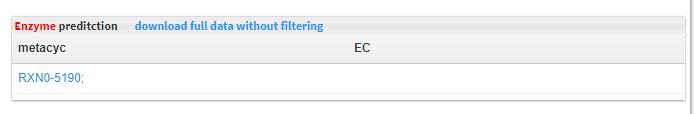
- 5.4 Enzyme prediction Ensemble Enzyme Prediction Pipeline annotates protein sequences with Enzyme Function classes comprised of full, four-part Enzyme Commission numbers and MetaCyc reaction identifiers.
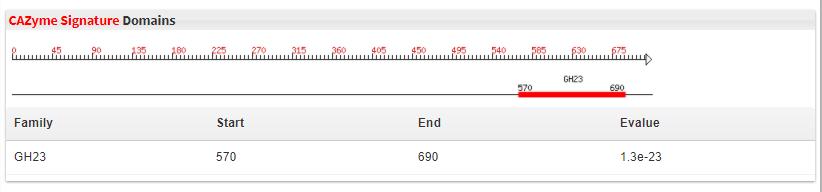
- 5.5 CAZyme Signature Domains CAZyme Signature Domains These are the family cazyme domains, the sequence was labled by red rectangles, which are different doamins.
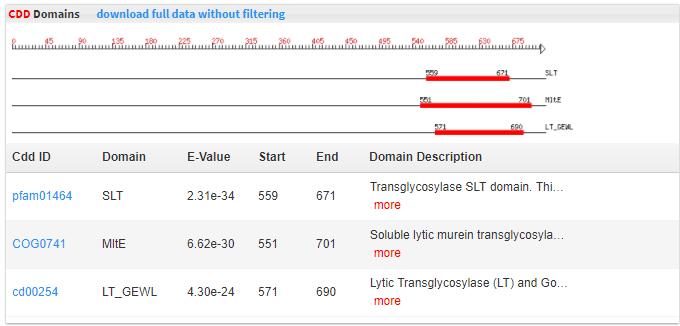
- 5.6 CDD search RPS-BLASTwas run with full-length CAZyme protein sequences as query and the NCBI CDD database (hyperlink) as the database. CDD is a protein annotation resource that contains well annotated sequence models. E-value < 1e-2 was used to keep the CDD domain match.
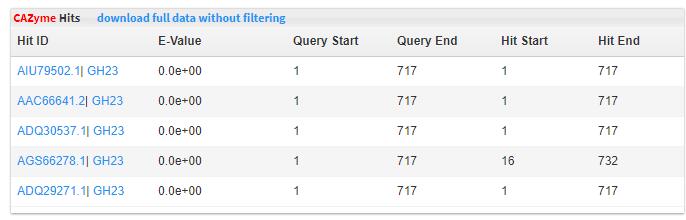
- 5.7 CAZyme Hits We use the Diamond search against DBCAN CAZyme Database instead of blast because of its fast search speed.
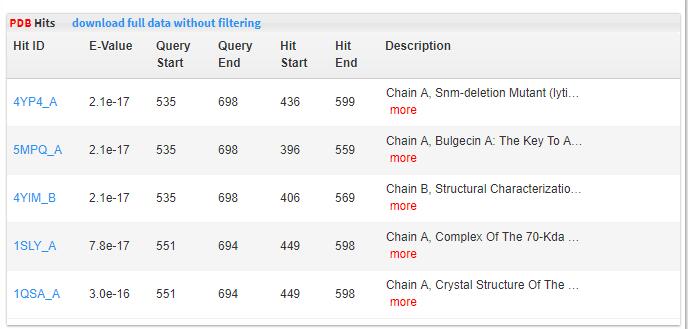
- 5.8 PDB hits Protein Data Bank protein sequence database was downloaded. Diamond was run with full-length proteins as query to search for homologous protein matches. Value <10 (E-value< 1e-5)was used to keep significant match. If there is a significant PDB match, that means the browsed protein has a close homolog with 3D structure solved. Orthologous groups.
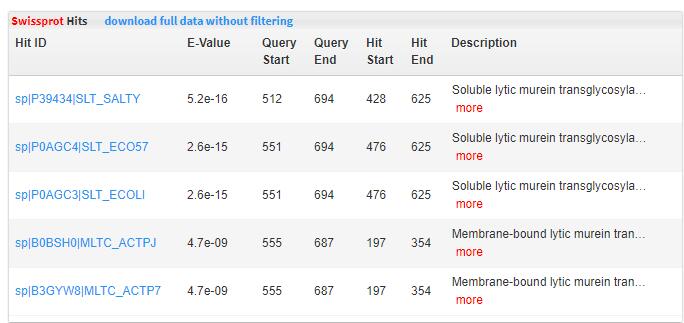
- 5.9 Swissprot hits Swiss-Prot database was downloaded. Diamond was run with full-length proteins as query to search for homologous protein matches. Value <10 (E-value< 1e-5)was used to keep significant match. If there is a significant PDB match, that means the browsed protein has a close homolog with 3D structure solved. Orthologous groups.
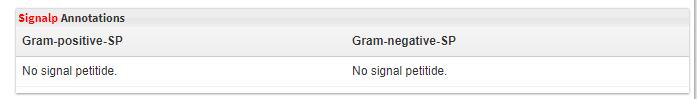
- 5.10 Signalp annotations Swiss-Prot database was downloaded. Signal peptide was predicted using SignalP. Both Gram-negative bacteria and Gram-positive bacteria are selected and their predicted results are offerd on the page(Gram-positve-SP and Gram-negative-SP).
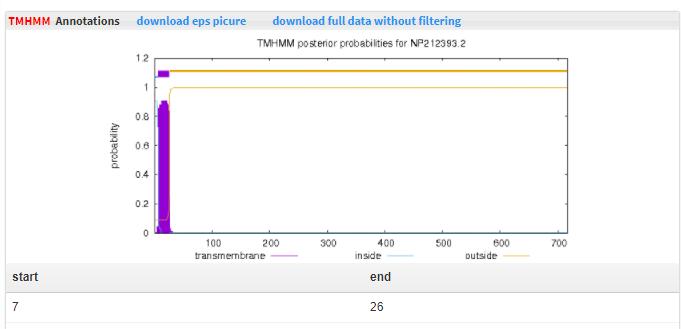
- 5.11 TMHMM Full-length sequences were taken to run TMHMMto predict the transmembrance regions.
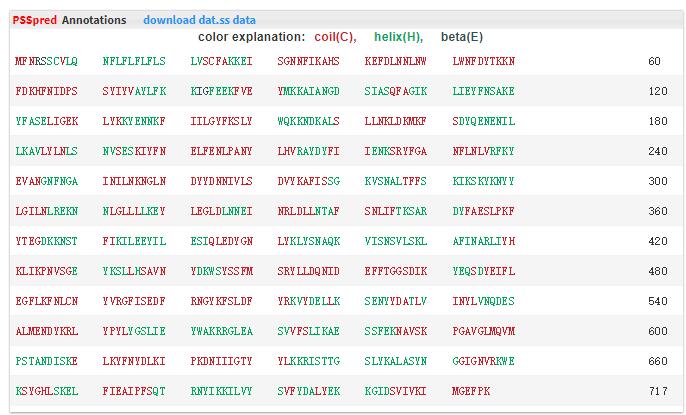
- 5.12 PPSpred The secondary structure prediction was predicted by PPSpred.
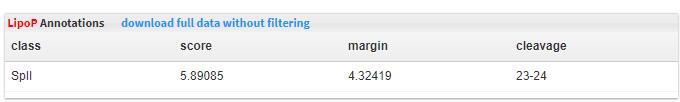
- 5.13 lipoP The lipoproteins prediction was predicted by lipoP.
We use different color(red - coil, green - helix, black - beta) to represent the different location.