1. Introduction (more details are in README)
pHMM-tree web server can flexibly handle four kinds of input: (1) multiple files each in pHMM format; (2) multiple files each with a multiple sequence alignment (MSA) in FASTA format; (3) mixture of 1 and 2; and (4) a file with unaligned protein sequences in FASTA format.
If the input is (1), all the pHMMs will be compared with each other using the pHMM comparison tool PRC (Madera, 2008) or HHsuite (Remmert, et al., 2012) to calculate a pair-wise distance. These distances will be then used to build a pHMM distance matrix, which will be used to build a distance-based phylogeny. If the input is (2), each MSA will be used to build a pHMM first and then build the phylogeny. If the input is (3), the unaligned sequences will be clustered into subfamilies according to some sequence identity threshold, and then each cluster will be aligned and all aligned clusters together will be used for building a pHMM phylogeny. Note that pHMM-tree is not a protein subclassification tool and the third type input will just do a simple similarity-based clustering and each cluster is treated as a subfamily. For specialized protein family subclassification tools, please see SCI-PHY and GeMMA.
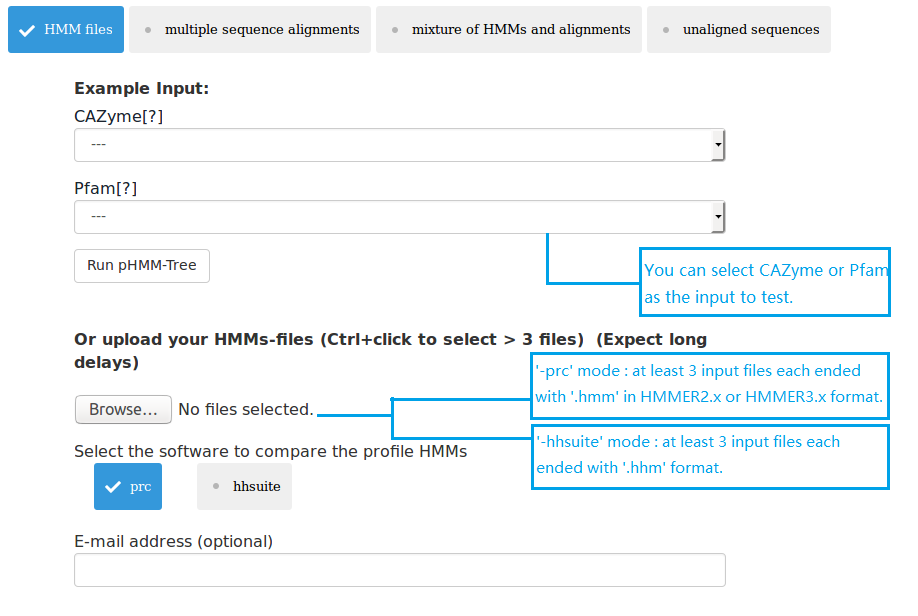
Standalone program: The pHMM-tree program was written in C/C++. It can take four kinds of input data as described above. Users should ensure that enough sequences, MSAs or pHMMs are provided as the inputs, because it needs at least three pHMMs to build a phylogeny. In addition to the parameter specifying the input data type, there are also parameters to (i) select one of the two different methods for pHMM comparison: PRC or HHsuite; (ii) change sequence identity thresholds for clustering. The output is written to a folder with at least three subfolders: (i) phylogeny; (ii) distance matrix; (iii) pHMM.
Web Server: In addition to the standalone program, we also developed a web server for users who do not have programming experience.
2. Web Interface
2.1 Prepare input data
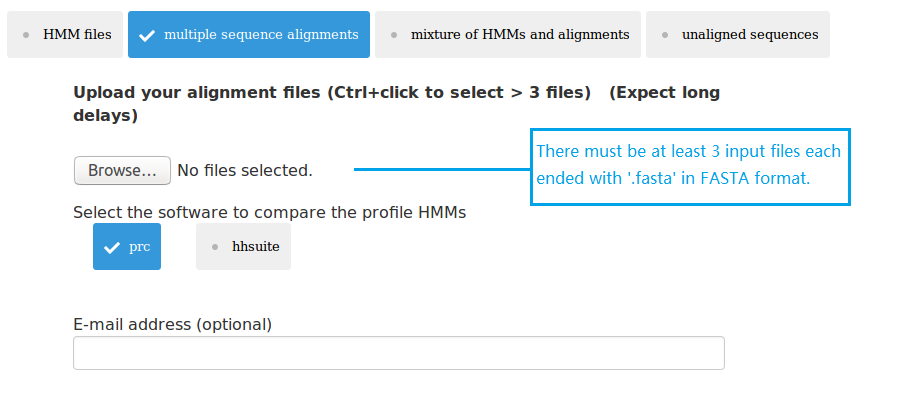
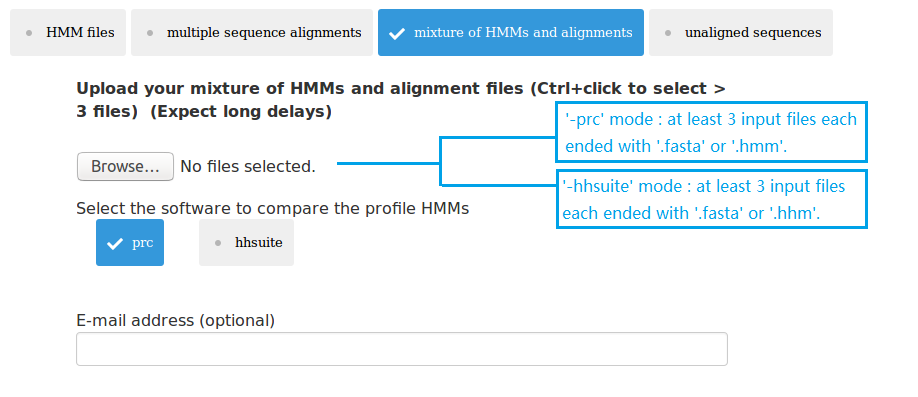
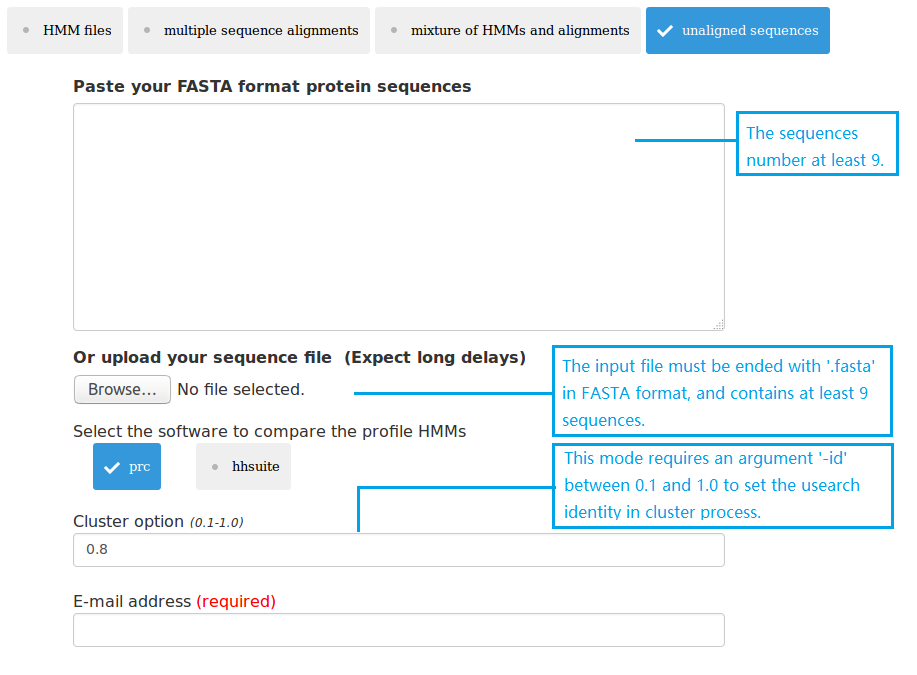
Three types of data can be submitted to pHMM-tree web server as input data. Therefore there are four input modes: (1) upload at least 3 HMM files; (2) upload at least 3 MSA files; (3) upload mixture of HMM and MSA files; (4) upload 1 single fasta file. The HMMs can be built using the HMMER package. The MSAs can be built using MAFFT or MUSCLE. The single fasta file must have enough sequences in it, so that after clustering using UCLUST there will be at least 3 cluster files each having at least 3 sequences.
2.2 Submit a Job
(1) upload at least 3 HMM files;
(2) upload at least 3 MSA files;
(3) upload at least 3 MSA or HMM files;
(4) upload 1 single fasta file;
2.3 Result page
The result page provides download links to newick format tree files using 6 different Phylip distance programs and tree graphs in PDF and PNG formats, as well as a zip file with all output files.