You are browsing environment: HUMAN GUT
Help page
You are here: Help
Index
There are several ways to browse through the available CAZymes and CGC and reach the annotation page for each individual CAZyme/CGC:
(i) browse by substrate via the tables/barplots on the Home page or Statistics pages.
(ii) browse by taxonomic rank, either by phylum using the tables/barplots on the Home page or Statistics
pages.
(iii) browse by PUL ID using the barplots on the Statistics pages.
(iv) browse and sort CGCs through all genomes on the CAZyme gene clusters page
(iv) search CAZyme or CGC by keywords including genome ID/CAZyme ID/CAZyme domain/CGC ID.

Navigation bar
The navigation contains Home, Download, CAZyme Gene Cluster, Statistics, Links, Help and About Us.
Download
 Clicking the Download
button on the navigation bar redirects users to a directory where all data for dbCAN-seq
available for download as well as additional files. The data is organized by environment.
each environment contain results:
Clicking the Download
button on the navigation bar redirects users to a directory where all data for dbCAN-seq
available for download as well as additional files. The data is organized by environment.
each environment contain results:
 Statistics
The Statistics page includes extended versions of the barplots featured on the CGCs.
These barplots visualize the breadth of types of substrates predicted by two approaches, phylum and homologous PULs.
The barplots can also be used for filtering and viewing CGGs by matching substrate, phylum or PULID. Users can click on a bar from any graph and be redirected to view CGCs page matching term.
Statistics
The Statistics page includes extended versions of the barplots featured on the CGCs.
These barplots visualize the breadth of types of substrates predicted by two approaches, phylum and homologous PULs.
The barplots can also be used for filtering and viewing CGGs by matching substrate, phylum or PULID. Users can click on a bar from any graph and be redirected to view CGCs page matching term.

Clicking the Download
button on the navigation bar redirects users to a directory where all data for dbCAN-seq
available for download as well as additional files. The data is organized by environment.
each environment contain results:
- dbCAN_overview.tar.gz All the overview from run_dbcan (one genome per folder in the tarball file).
- cgc_result.tar.gz All the cgc result from run_dbcan (one genome per folder in the tarball file).
- cazyme.fa.tar.gz All the faa of cazymes (one genome per folder in the tarball file).
- cgc.fa.tar.gz All the faa of cgc (one genome per folder in the tarball file).
- substrates.tar.gz the substrate table, each substrate per file in the tarball. Each column is seperated by tab. The column name for each substrate file is:
The fasta sequence ID was named in format: ContigID|CGCorder|proteinID|Type. Example: MGYG000291367_10|CGC1|MGYG000291367_00645|CAZyme
CGC ID was named with ContigID and CGCorder. The CGC ID for the example CGC fasta sequences is MGYG000291367_10|CGC1
geneomeID CGCID CGC_content PULDID substrate species_name method signature_content
Statistics
The Statistics page includes extended versions of the barplots featured on the CGCs.
These barplots visualize the breadth of types of substrates predicted by two approaches, phylum and homologous PULs.
The barplots can also be used for filtering and viewing CGGs by matching substrate, phylum or PULID. Users can click on a bar from any graph and be redirected to view CGCs page matching term.
Search engine
Introduction: A search box is shown at the top part of all pages.
The search function is built upon the powerful search engine Sphinx,
which has been programmatically configured to allow very fast index-organized table search
(average response time: 300 millisecond) and highly efficient pagination.
It implements a Google-like search supporting both exact and fuzzy query, and users
can input a keyword to search 5 different data types.
1.1 Searching CAZymes by using CAZyme ID as keyword.
1.2 Searching CAZymes by using genome ID as keyword.
1.3 Searching CAZymes by using species name as keyword.
1.4 Searching CAZymes by using CAZyme family as keyword.
1.5 Searching CGCs by using substrate as keyword.
Examples:
2.1 CAZyme ID (e.g MGYG000000001_00067 )
2.2 CAZyme IDs (e.g MGYG000000001_00067|MGYG000004854_00178 )
2.3 Genome ID (e.g MGYG000000001 )
2.4 Speice name (e.g Cloacibacillus porcorum )
2.5 CAZyme family (e.g CE4)
2.6 Substrate (e.g beta-glucan )
1.1 Searching CAZymes by using CAZyme ID as keyword.
1.2 Searching CAZymes by using genome ID as keyword.
1.3 Searching CAZymes by using species name as keyword.
1.4 Searching CAZymes by using CAZyme family as keyword.
1.5 Searching CGCs by using substrate as keyword.
Examples:
2.1 CAZyme ID (e.g MGYG000000001_00067 )
2.2 CAZyme IDs (e.g MGYG000000001_00067|MGYG000004854_00178 )
2.3 Genome ID (e.g MGYG000000001 )
2.4 Speice name (e.g Cloacibacillus porcorum )
2.5 CAZyme family (e.g CE4)
2.6 Substrate (e.g beta-glucan )
Four environments and dbCAN-seq refseq
We included four MAG datasets from the EBI MGnify database.
Each dataset contains genomes of thousands of prokaryotic species, and each species often have multiple MAGs.
1. The human gut dataset (also known as the Unified Human Gastrointestinal Genome [UHGG] collection) contains 4,744 species.
2. The human oral dataset contains 452 species.
3. The cow rumen dataset contains 2,729 species.
4. The marine database contains 1.496 species.
By clicking the specify icons on the main page, users can access the CAZymes and CGCs from different dataset.
1. The human gut dataset (also known as the Unified Human Gastrointestinal Genome [UHGG] collection) contains 4,744 species.
2. The human oral dataset contains 452 species.
3. The cow rumen dataset contains 2,729 species.
4. The marine database contains 1.496 species.
By clicking the specify icons on the main page, users can access the CAZymes and CGCs from different dataset.
Broswe CAZymes by taxonomy
We group all the CAZymes by phylum and show the number of species in the specify phylum.
By clicking the phylum in the main page, taxonomic group and geneome bar are shown on the page.
Following the instruction in the figure, you can broswe the CAZyme by taxonomy.

Broswe CAZymes by family
We group all the CAZymes by CAZyme family and show the number of CZyme in each family.
Following the instruction in the figure, you can broswe the CAZyme by taxonomy.

Two substrate prediction approaches
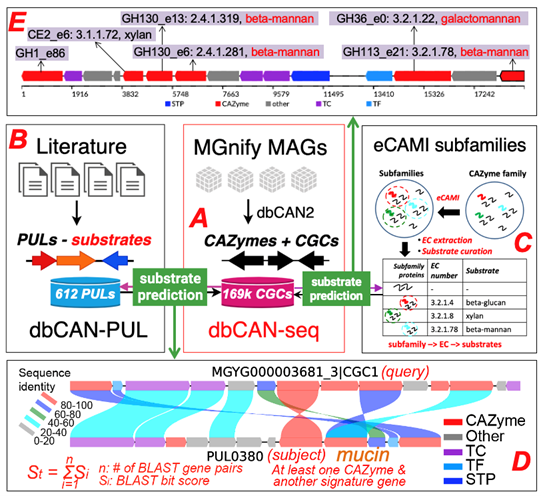 Two approaches are employed to predict the substrates of the CGCs.
Two approaches are employed to predict the substrates of the CGCs.
1.1 dbCAN-PUL search.
This approach is based on the BLAST comparison of protein sequences of the query CGCs against the protein sequences of the 612 PULs of dbCAN-PUL. To improve the confient of the CGC predict, all the CGC hits are sorted by the summed bit-scores of each genes. In addtion, it requires at least one CAZyme match plus at less one match from one of the other 4 signature genes.
1.2. eCAMI sub-family.
This pproach is based on the inspection of the eCAMI subfamily annotated substrates of all component CAZymes in the CGC. The substate of CGC is dependent on the substrate of CAZymes in the CGC. It requires at less two shared two CAZyme substrate to obtain a higerh confident substrate for the CGC. The final substrate is decided as the most common shared CAZyme substrate. Broswe CAZymes by substrate We group all the CGCs by the substrate and show the number of CGC in the specify substrate. Following the instruction in the figure, you can broswe the CAZyme by taxonomy.

Two approaches are employed to predict the substrates of the CGCs. 1.1 dbCAN-PUL search.
This approach is based on the BLAST comparison of protein sequences of the query CGCs against the protein sequences of the 612 PULs of dbCAN-PUL. To improve the confient of the CGC predict, all the CGC hits are sorted by the summed bit-scores of each genes. In addtion, it requires at least one CAZyme match plus at less one match from one of the other 4 signature genes.
1.2. eCAMI sub-family.
This pproach is based on the inspection of the eCAMI subfamily annotated substrates of all component CAZymes in the CGC. The substate of CGC is dependent on the substrate of CAZymes in the CGC. It requires at less two shared two CAZyme substrate to obtain a higerh confident substrate for the CGC. The final substrate is decided as the most common shared CAZyme substrate. Broswe CAZymes by substrate We group all the CGCs by the substrate and show the number of CGC in the specify substrate. Following the instruction in the figure, you can broswe the CAZyme by taxonomy.
Basic Information
The basic information descibes the properties of CAZyme and its genome.
Some of them have the external links to other databases.
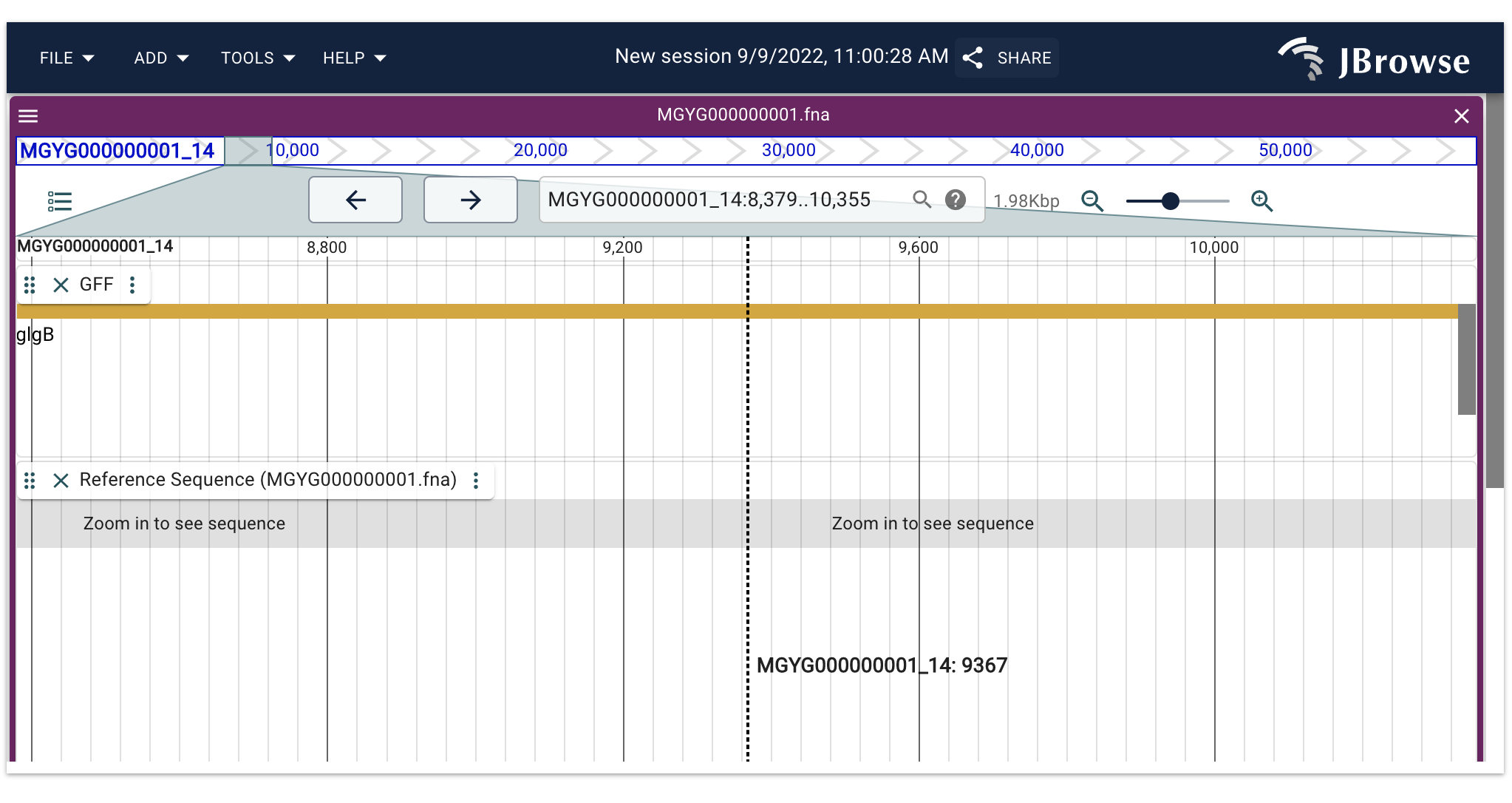
 Genomic Context
Jbrowse2 uses the GFF3 file to display the genomic location of the gene and its neighboring genes on the chromosome.
Genomic Context
Jbrowse2 uses the GFF3 file to display the genomic location of the gene and its neighboring genes on the chromosome.
 Full sequence
We provide the full-length sequence of the protein and a download link.
Full sequence
We provide the full-length sequence of the protein and a download link.
 Enzyme prediction
eCAMI annotates protein sequences with Enzyme Function classes (EC numbers).
Enzyme prediction
eCAMI annotates protein sequences with Enzyme Function classes (EC numbers).
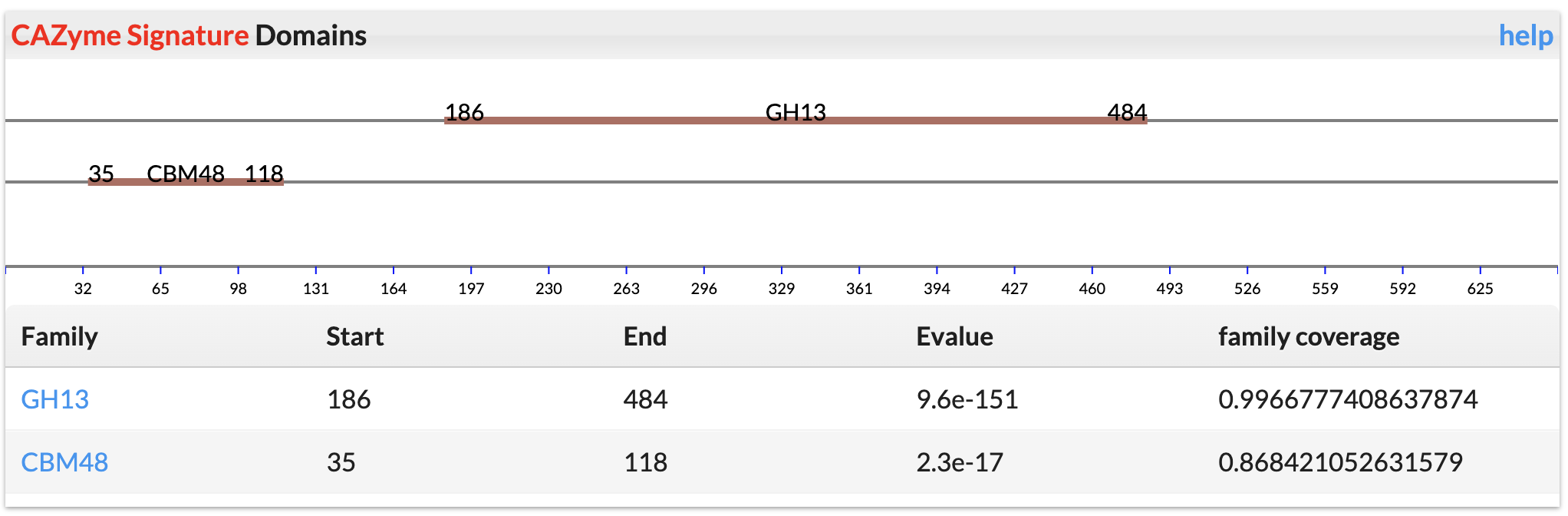
 CAZyme Signature Domains
These are CAZyme domains annotated by dbCAN.
CAZyme Signature Domains
These are CAZyme domains annotated by dbCAN.
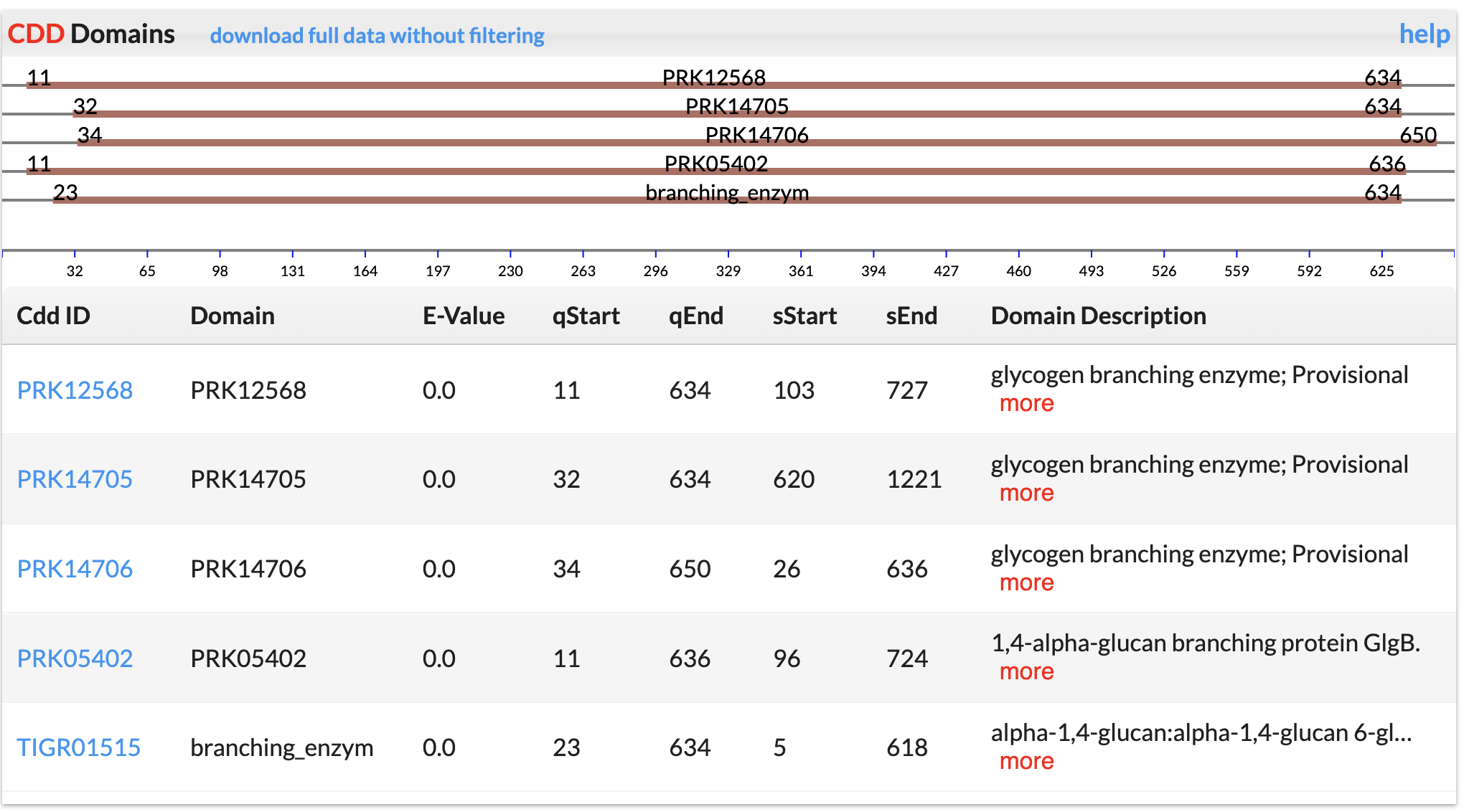
 CDD domain
RPS-BLAST was run with full-length CAZyme protein sequences as query and the NCBI CDD database as the database.
CDD is a protein annotation resource that contains well annotated sequence models.
E-value < 1e-2 was used to keep the CDD domain hits.
CDD domain
RPS-BLAST was run with full-length CAZyme protein sequences as query and the NCBI CDD database as the database.
CDD is a protein annotation resource that contains well annotated sequence models.
E-value < 1e-2 was used to keep the CDD domain hits.
 CAZyme Hits
We use the DIAMOND program to search against the CAZy annotated CAZyme sequences
CAZyme Hits
We use the DIAMOND program to search against the CAZy annotated CAZyme sequences
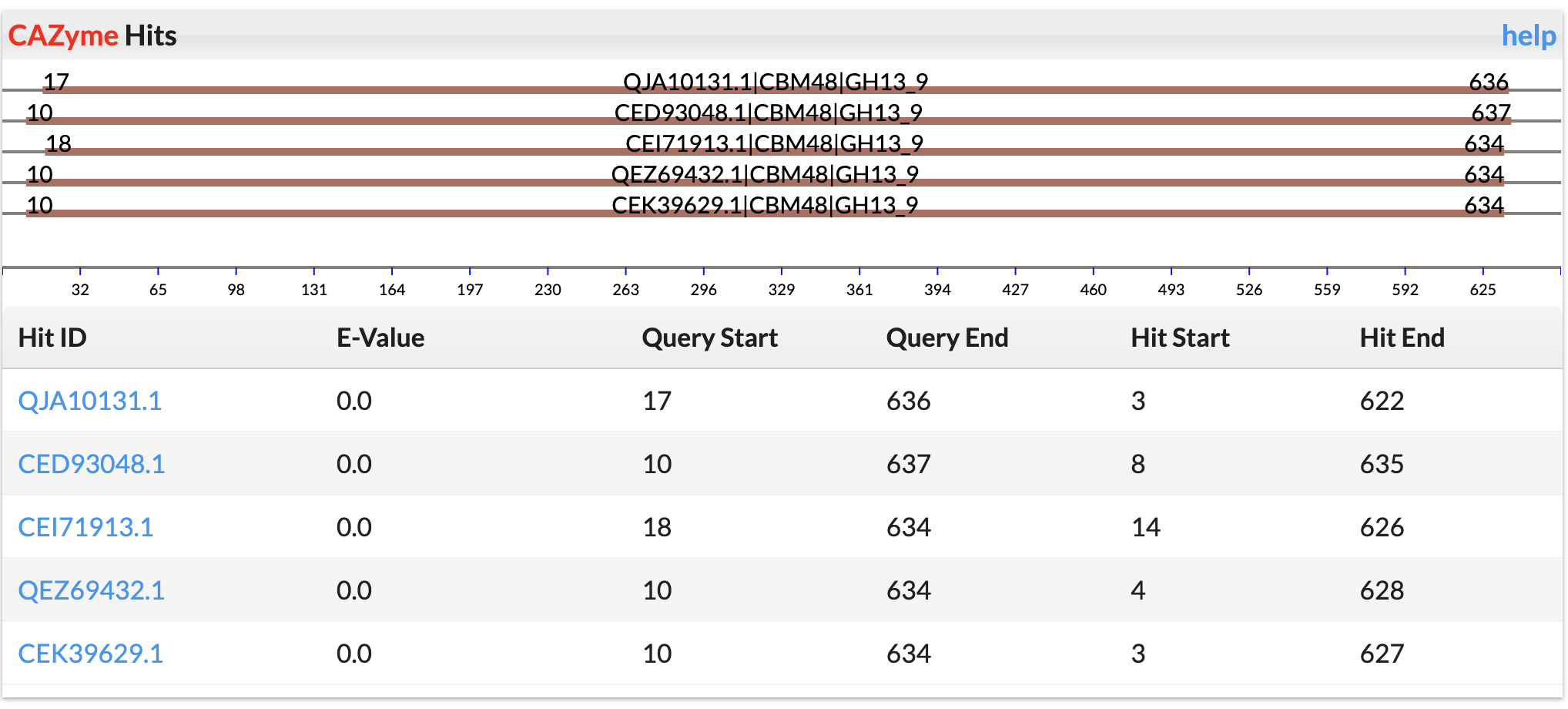 PDB Hits
The Protein Data Bank protein sequences was downloaded and searched against with DIAMOND program.
E-value < 1e-5 was used to keep significant hits.
PDB Hits
The Protein Data Bank protein sequences was downloaded and searched against with DIAMOND program.
E-value < 1e-5 was used to keep significant hits.
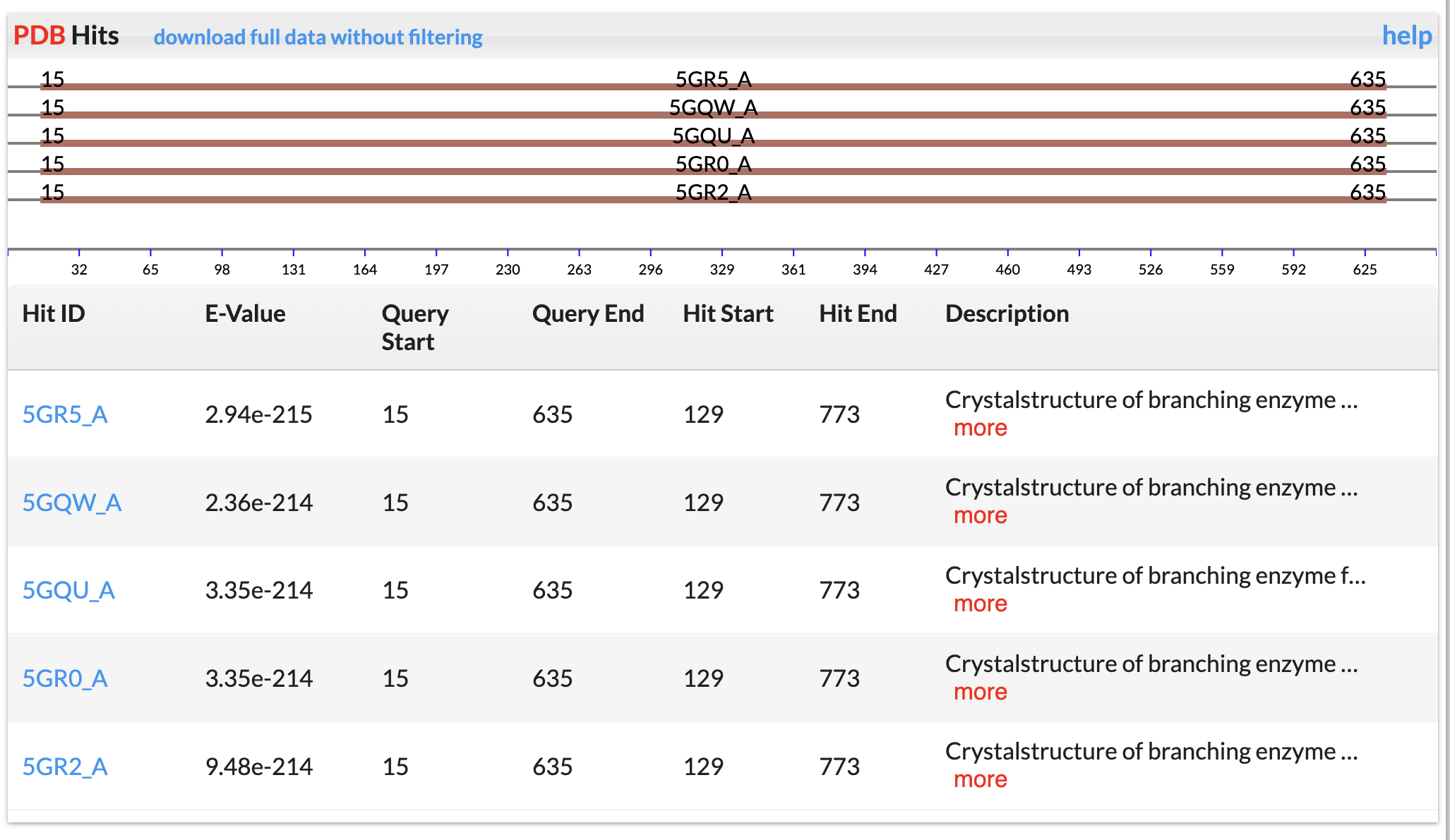 Swiss-Prot Hits
Swiss-Prot database was downloaded. E-value < 1e-5 was used to keep significant hits.
Swiss-Prot Hits
Swiss-Prot database was downloaded. E-value < 1e-5 was used to keep significant hits.
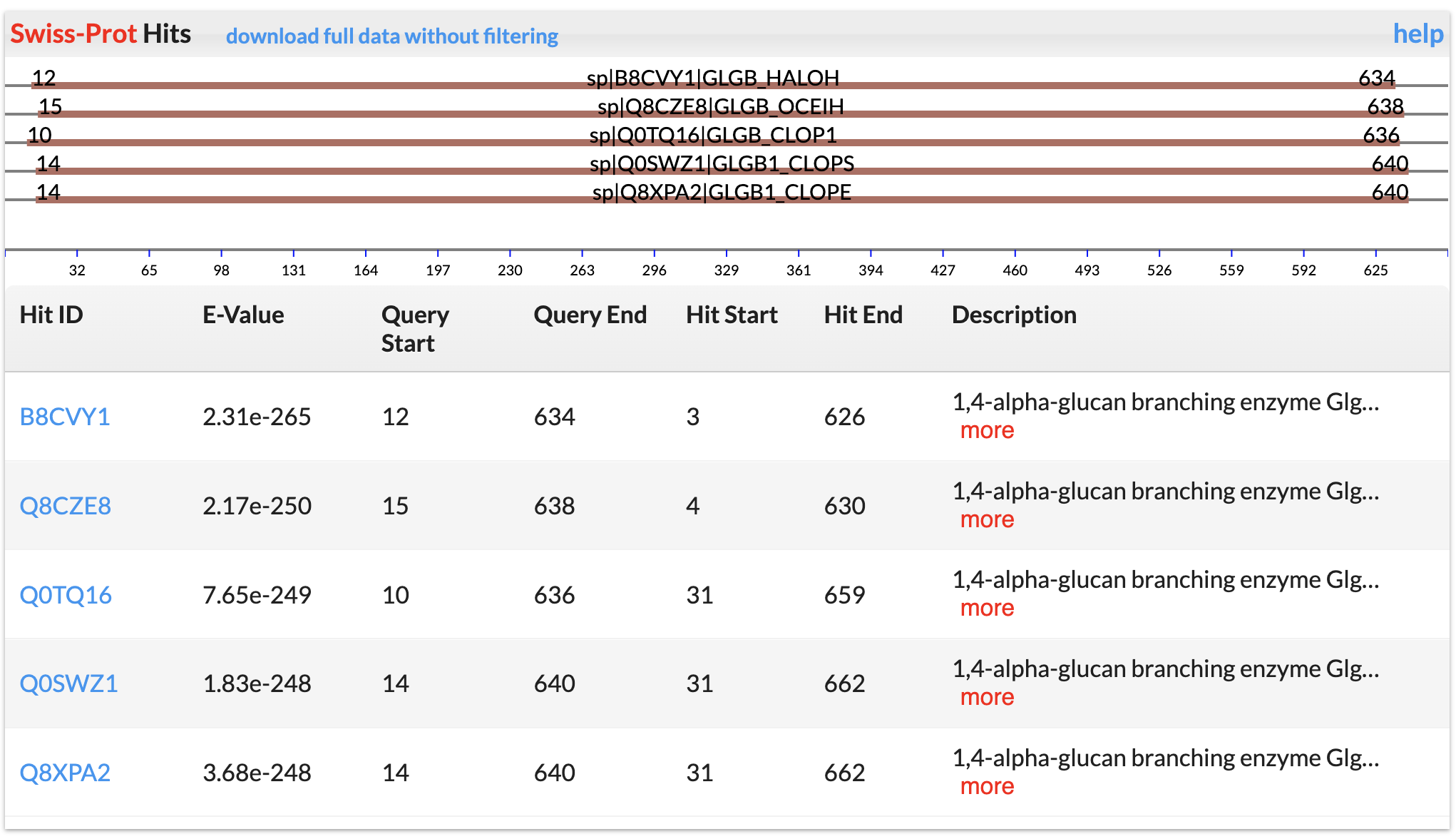 SignalP and LipoP Annotations
Signal peptide and lipoprotein were predicted using SignalP.
SignalP and LipoP Annotations
Signal peptide and lipoprotein were predicted using SignalP.
 TMHMM annotation
Full-length sequences were taken to run TMHMMto predict the transmembrane regions.
TMHMM annotation
Full-length sequences were taken to run TMHMMto predict the transmembrane regions.

Genomic Context
Jbrowse2 uses the GFF3 file to display the genomic location of the gene and its neighboring genes on the chromosome.
Full sequence
We provide the full-length sequence of the protein and a download link.
Enzyme prediction
eCAMI annotates protein sequences with Enzyme Function classes (EC numbers).
CAZyme Signature Domains
These are CAZyme domains annotated by dbCAN.
CDD domain
RPS-BLAST was run with full-length CAZyme protein sequences as query and the NCBI CDD database as the database.
CDD is a protein annotation resource that contains well annotated sequence models.
E-value < 1e-2 was used to keep the CDD domain hits.
CAZyme Hits
We use the DIAMOND program to search against the CAZy annotated CAZyme sequences
PDB Hits
The Protein Data Bank protein sequences was downloaded and searched against with DIAMOND program.
E-value < 1e-5 was used to keep significant hits.
Swiss-Prot Hits
Swiss-Prot database was downloaded. E-value < 1e-5 was used to keep significant hits.
SignalP and LipoP Annotations
Signal peptide and lipoprotein were predicted using SignalP.
TMHMM annotation
Full-length sequences were taken to run TMHMMto predict the transmembrane regions.
CGC gene composition diagram
The gene cluster strcture will be depicted at CGC Page with putative CGC signature genes highlighted by predicted function:
TC = Transporter (purple color gene)
TF = transcription factor (light blue color gene)
STP = signal transduction protein (dark blue color gene)
CAZyme = Carbohydrate Active Enzyme (red color gene)
null = non-signature gene (grey color gene)
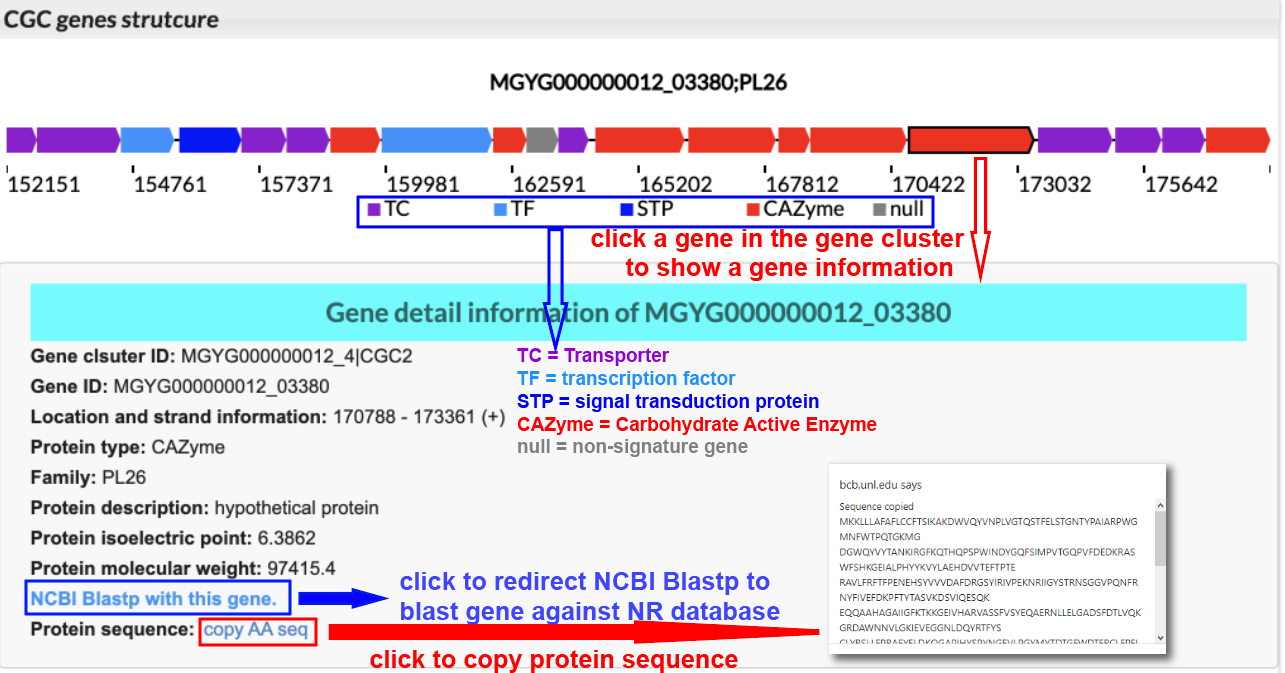 CGC gene composition table
CGC gene composition table
 Substrate predicted by dbCAN-PUL search
Substrate predicted by dbCAN-PUL search
 Substrate predicted by eCAMI subfamily
Substrate predicted by eCAMI subfamily
 Genomic Context
Genomic Context

CGC gene composition table
Substrate predicted by dbCAN-PUL search
Substrate predicted by eCAMI subfamily
Genomic Context
Copyright 2022 © YIN LAB, UNL. All rights reserved. Designed by Jinfang Zheng and Boyang Hu. Maintained by Yanbin Yin.